
ABSTRACT
Introduction: Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disease of skin and mucous membranes. EBA is caused by autoantibodies against type VII collagen, which is a major component of anchoring fibrils, attaching epidermis to dermis. Binding of autoantibodies to type VII collagen leads to skin fragility and, finally, blister formation. The clinical picture of EBA is polymorphic, with several distinct phenotypes being described. Despite recent progress in understanding the pathophysiology of EBA, its diagnosis is still challenging.
Areas covered: This review provides an update on the clinical manifestations and diagnostic methods of EBA. We searched PubMed using the terms ‘epidermolysis bullosa acquisita’ covering articles in English between 1 January 2005 and 31 May 2016. Relevant older publications were retrieved form cited literature.
Expert commentary: While the clinical picture is highly variable, diagnosis relies on direct immunofluorescence (IF) microscopy of a perilesional skin biopsy. Linear deposits of IgG, IgA and/or C3 along the dermal-epidermal junction with an u-serrated pattern are diagnostic for EBA alike the detection of serum autoantibodies against type VII collagen. Several test systems for the serological diagnosis of EBA have recently become widely available. In some patients, sophisticated diagnostic approaches only available in specialized centers are required.
1. Introduction
In the past decades, the incidence of autoimmune diseases in developed countries is increasing resulting in a major health burden [Citation1]. Autoimmune bullous diseases (AIBD) are a heterogeneous group and one of the major organ-specific autoimmune diseases.
AIBD can be separated in three different groups, namely pemphigoid diseases, pemphigus diseases, and dermatitis herpetiformis. Pemphigoid diseases are characterized by the presence of autoantibodies against structural proteins of dermal–epidermal junction and lead to subepidermal blistering, whereas in pemphigus diseases, autoantibodies against desmosomal proteins are detected causing intraepidermal blister formation. Dermatitis herpetiformis is always associated with celiac disease and characterized by autoantibodies against transglutaminase 2 and 3 [Citation2–Citation4].
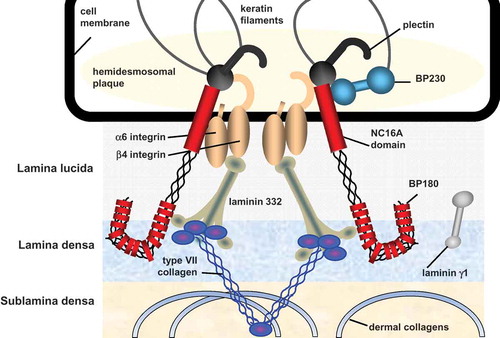
Epidermolysis bullosa acquisita (EBA) is a rare chronic autoimmune blistering disease of skin and mucous membranes characterized by circulating and tissue bound antibodies against type VII collagen [Citation5,Citation6]. Type VII collagen is the major component of anchoring fibrils, which provides attachment of the epidermis and underlying basement membrane zone (BMZ) to the dermis () [Citation7–Citation9]. Destruction of anchoring fibrils leads to detachment of the epidermis from the dermis resulting in skin fragility, blister formation, erosions that frequently heal with scarring and milia formation. Clinical presentations of EBA are heterogeneous. Two major clinical variants have been described, the classical mechanobullous form [Citation10] and the inflammatory variant [Citation11]. Within latter form, subtypes resembling bullous pemphigoid, mucous membrane pemphigoid, linear immunoglobulin A (IgA) disease, and Brunsting–Perry pemphigoid can be differentiated [Citation11–Citation14]. Diagnosis of EBA can, thus, not be based on the clinical picture alone but requires a combination of laboratory investigations including direct immunofluorescence (IF) microscopy and serological assays. In this review, we will provide an update on the clinical manifestations and diagnostic options of EBA and will shortly summarize recent epidemiological and pathophysiological discoveries of this disease.
Figure 1. Schematic structure of the dermal-epidermal junction. Only proteins that are targeted by autoantibodies in autoimmune blistering diseases are depicted.
2. History
EBA was initially described by G.T. Elliott in 1895 in two patients clinically reminiscent of epidermolysis bullosa hereditaria but with no affected family members [Citation15]. In 1971, Henry H. Roenigk established the first diagnostic criteria for the mechanobullous form of EBA, i.e. (1) onset of the disease in adulthood, (2) absence of familial history for bullous diseases, (3) trauma-induced or spontaneous formation of blisters resembling dystrophic epidermolysis bullosa, and (4) exclusion of other bullous diseases. In 1984, David Woodley identified the target antigen of EBA as a 290-kDa protein in an extract of human dermis [Citation16]. This 290-kDa protein was later shown to be type VII collagen [Citation17].
Traditionally, EBA has been classified as an entity outside the pemphigoid group defining pemphigoid diseases as disorders with hemidesmosomal target antigens [Citation18]. For dermatologists not primarily involved in the care of AIBD patients as well as other physicians, these subtle differences may hardly be appreciated. We therefore advocate to classify pemphigoid diseases based on reactivity against structural proteins of the dermal–epidermal junction rather that against hemidesmosomal proteins [Citation3].
3. Epidemiology
EBA is one of the rarest autoimmune blistering diseases with an incidence of 0.2–0.5/million inhabitants/year [Citation19–Citation23]. In a recent analysis of data from the largest German insurance company from 2014, the prevalence of EBA was calculated to be 2.84/million inhabitants, i.e. about 230 EBA patients in Germany [Citation24].
The disease can affect all age groups with an average onset of disease at the age of 50 years [Citation25–Citation27]. Children and elderly can also be affected [Citation28–Citation30] with children making up about 10% of EBA patients (unpublished observation). No gender predisposition is known [Citation24,Citation26,Citation27,Citation31].
In two studies, EBA occurred more frequently in black patients of African descent [Citation25,Citation32]. EBA is associated with the HLA-DR2 (corresponding to HLA-DRB1*15) and DRB1*15:03, an allele found frequently in the general population [Citation25,Citation32]. Korean EBA patients more frequently carried DRB1*13 compared to controls [Citation33]. This finding of an association with the major histocompatibility complex locus is paralleled in a mouse model of the disease, where susceptibility to develop clinical EBA lesions after manifestations is closely linked to the H2s haplotype [Citation34].
While in 25% and 16% of patients from USA and France, an association with inflammatory bowel disease was observed this association was not found in Korean patients [Citation25,Citation27,Citation35,Citation36]. A variety of other diseases, e.g. rheumatoid arthritis, diabetes, cryoglobulinemia, and psoriasis were reported in individual EBA patients [Citation5,Citation37,Citation38]. Recently, an association with hematological malignancies i.e. lymphoma was observed in about 8% of EBA patients [Citation39] in line with a previous study with 102 AIBD patients [Citation40].
As triggering factors of EBA, penicillin, vancomycin, gentamycin, UV-radiation, and contact allergy to metals have been reported [Citation41–Citation44].
4. Pathophysiology
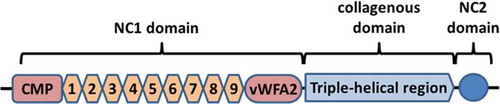
Type VII collagen is composed of three identical alpha-chains each consisting of a 145-kDa N-terminal non-collagenous (NC)-1 domain, a large central collagenous domain, and a smaller C-terminal NC2 domain [Citation7]. Type VII collagen molecules form antiparallel tail-to-tail dimers with the help of the NC2 domains that are subsequently removed during this process [Citation7,Citation45]. The NC1 domain consists of N-terminal protein with homology to cartilage matrix protein, nine fibronectin-like domains, and C-terminal portion with homology to the von Willebrand factor A domain (). The NC1 domain interacts with several structural molecules of the BMZ such as type IV collagen, laminin 332, fibronectin, and type I dermal collagen [Citation46–Citation50]. The NC1 domain was previously described as major antigenic portion in EBA [Citation51–Citation53], however, patients with antibodies against the NC2 and the collagenous domains have been described [Citation54–Citation59]. Perturbation of the interaction of the NC1 domain with these molecules caused by binding of autoantibodies against specific subdomains of NC1 is discussed to explain the variation of in the clinical phenotypes of EBA. However, to this moment, no clear correlation between antitype VII collagen autoantibody specificities and a distinct clinical phenotype has been described [Citation60,Citation61]. Another explanation of blister formation could be the direct interference with the NC2 domain and hindering of antiparallel tail-to-tail dimer formation [Citation62].
Figure 2. Structure of type VII collagen. CMP: cartillage matrix protein; 1–9: fibronectin III-like repeats 1–9; vWFA2: von Willebrand factor A; NC: noncollagenous.
Detailed analyses of the autoantibody response in EBA revealed the presence of IgG in the majority of patients with predominance of the IgG1 and IgG4 subclasses [Citation63–Citation65]. In addition, IgA antitype VII collagen reactivity, either exclusively or in combination with IgG autoantibodies, is observed in 50–60% of patients [Citation26,Citation27,Citation66].
The pathogenic relevance of antibodies against type VII collagen has clearly been shown in several in vivo and in vitro models [Citation67–Citation71] reviewed in [Citation6]. Incubation of cryosections of normal human skin with sera of EBA patients followed by incubation with neutrophils from healthy volunteers leads to dermal–epidermal split formation [Citation69]. Injection of antitype VII collagen antibodies in mice results in a skin disease that clinically and immunopathologically mimics the human disease [Citation61,Citation67,Citation71–Citation73]. While these models well reflect characteristic features of human EBA, they only reproduce the effector phase of EBA, i.e. the antitype VII collagen-mediated tissue destruction. To investigate the loss of tolerance to type VII collagen, mice were immunized with recombinant fragments of NC1 domain, which raised murine antitype VII collagen IgG that resulted in a long-lasting autoimmune skin disease resembling human EBA [Citation68,Citation74]. These animal models were subsequently used to show the pathogenic relevance of complement activation at the epidermal BMZ, neutrophils, CD18-mediated extravasation, FcγRIV, heat-shock protein 90, the glycosylation status of antitype VII collagen IgG, IL-1β, IL-6, GM-CSF, Erk 1/2, CXCL1/2, p38, Akt, RORa, HSP90, gene expression in myeloid cells, and the skin microbiome [Citation75–Citation84].
5. Clinical presentation
Five different phenotypes of EBA have been reported so far, including the classical mechanobullous variant described by Roenigk [Citation10] and the four inflammatory forms resembling bullous pemphigoid, mucous membrane pemphigoid, linear IgA-disease, and Brunsting–Perry pemphigoid, respectively [Citation11–Citation14]. About one-third of EBA patients present with the classical phenotype and about two-thirds with one of the inflammatory variants [Citation26,Citation27]. Clinical subtypes, however, are neither categorical nor static. Some patients present with mixed phenotypes and in others, the subtype changes during the course of their disease [Citation26]. Mucosal involvement is found in about 60% of patients but only predominates over skin lesions in about 5–10% of EBA, i.e. in those with the mucous membrane pemphigoid variant [Citation26,Citation27].
5.1. Mechanobullous variant
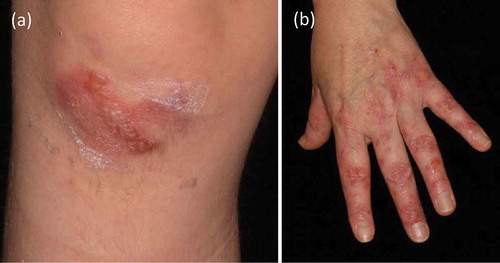
This classical form of EBA demonstrates skin fragility, vesicles, tense blisters, and erosions on non-inflamed skin, which heal with scarring and milia formation [Citation10]. Lesions can affect every region of skin and mucous membranes with predisposition to trauma-prone areas, such as elbows, knees, hands, feet, knuckles, toes, sacral area (). In the course of disease, cicatricial alopecia and onychodystrophy may appear. In mild forms of the mechanobullous variant, porphyria cutanea tarda and pseudoporphyria need to be excluded. Severe forms of classical EBA can imitate hereditary epidermolysis bullosa [Citation85].
Figure 3. Classical variant of EBA. (a) Erythema and tense blisters on the right knee of a 6-year old boy. B. Erythema and erosions on the left hand of a 53-year old female.
5.2. Bullous pemphigoid-like variant
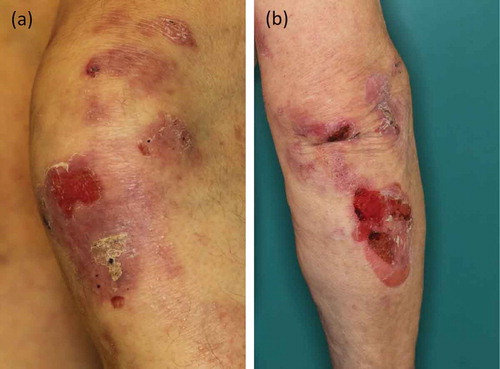
Bullous pemphigoid is by far the most frequent autoimmune blistering disease [Citation86–Citation88] (reviewed in [Citation89]). Bullous pemphigoid typically presents with tense blisters, erosions on inflamed or otherwise unaffected skin, crusts, and urticarial-like erythema [Citation90]. Like in bullous pemphigoid, in this EBA variant, pruritus is often reported, while milia and scar formation are absent (). Lesions mainly involve the trunk, skin folds, and extremities [Citation11,Citation91]. This inflammatory form of EBA is found in 25–50% of all reported EBA cases [Citation27].
Figure 4. Inflammatory variant of EBA. (a) Erythema, erosions, and crusts on the left knee of a 76-year old patient. (b) Blisters and erosions on the right elbow of the same EBA patient.
5.3. Mucous membrane pemphigoid-like variant
Mucous membrane pemphigoid is a pemphigoid disease characterized by the predominant or exclusive involvement of mucous membranes [Citation92]. All squamous cell epithelia can be affected, including oral and nasal mucosa, larynx, esophagus, anal and genital mucosa as well as conjunctiva [Citation14,Citation93–Citation95]. While 50–65% of EBA patients have mucosal lesions [Citation26,Citation96,Citation97] () in about 5–10% of patients, mucosal involvement predominates and patients may be classified as mucous membrane pemphigoid-variant of EBA [Citation27,Citation85]. In some EBA patients, exclusive involvement of the esophagus or nasal mucosa has been reported [Citation98]. In this variant, considerable overlap occurs with mucous membrane pemphigoid itself. Many physicians will according to the consensus conference on mucous membrane pemphigoid classify patients with autoantibodies against type VII collagen and predominant mucosal lesions as mucous membrane pemphigoid [Citation92], others as mucous membrane pemphigoid-like variant of EBA. When conjunctival or laryngeal lesions are present, the authors would recommend classification as mucous membrane pemphigoid since treatment may follow protocols for latter disease including more aggressive regimens, e.g. with cyclophosphamide [Citation92].
Figure 5. Mucosal involvement. (a) Blisters and erosions on the lower lip of a 6-year old boy (same patient as in . (b) Erosions on the tongue of a 76-year old male with EBA (same patient as in ,)
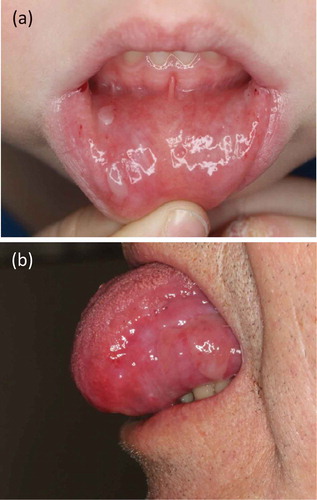
5.4. Linear IgA disease-like variant
This EBA variant presents with tense vesicles, blisters, erythema, and urticated plaques in an annular or polycyclic pattern with vesicles frequently localized along the edge of the lesion. This pattern is associated with (but not limited to) linear IgA dermatosis and is termed ‘string-of-pearls’ or ‘crown-of-jewels’ sign. Scarring and milia formation are absent like in linear IgA disease. Mucosal involvement, and in 4% of cases, severe conjunctival lesions have been described [Citation85,Citation99]. As in linear IgA disease, linear deposits of IgA are visualized by direct IF microscopy of a perilesional biopsy. It may be debated if these patients are classified as linear IgA disease with autoantibodies against type VII collagen or as linear IgA disease-like variant of EBA, also called IgA EBA [Citation100,Citation101]. Interestingly, in 11% of patients with this variant, a close relation to the initiation of a new drug has been reported [Citation102]. Otherwise, no difference between EBA patients with exclusive or predominant IgA reactivity compared to patients with IgG antitype VII collagen antibodies has been detected [Citation26].
5.5. Brunsting–Perry pemphigoid-like variant
This rare clinical form is characterized by subepidermal blisters, erosions, hemorrhagic crusts, and atrophic scars on the head and neck without involvement of mucous membranes [Citation103]. In most patients with Brunsting–Perry pemphigoid, antibodies against type VII collagen have been detected, thus classifying as Brunsting–Perry pemphigoid-like variant of EBA [Citation12,Citation26,Citation27,Citation104–Citation106].
In addition, a single patient with congenital EBA has been described [Citation107]. A mother with known EBA delivered an otherwise healthy girl with tense blisters and erosions. In the newborn, direct IF microscopy showed linear deposits of IgG and C3 at the BMZ and serum antibodies against type VII collagen were detected. Blistering ceased within 10 days under supportive care and no recurrence was noted [Citation107]. This case clearly demonstrates the pathogenic potential of type VII collagen-specific autoantibodies.
6. Diagnostic methods
Due to the variety of clinical presentations, diagnosing EBA based on the clinical picture alone is not possible. In case of clinical suspicion of EBA, diagnosis needs to be based on different approaches schematically shown in . Traditionally, the diagnostic gold standard has been direct immunogold electron microscopy [Citation108]. At present, in nearly all patients, diagnosis can be made by serration pattern analysis of direct IF microcopy or/and detection of serum autoantibodies against type VII collagen [Citation16,Citation109,Citation110]. For latter analysis, three commercial test systems are currently available [Citation55,Citation65].
6.1. Histopathology
Histopathology of a lesional skin biopsy allows distinguishing between intraepidermal (like in pemphigus) and subepidermal blistering characteristic for pemphigoid disorders such as EBA. In the classical mechanobullous form, a scarce or no inflammatory infiltrate in the dermis can be detected. In the inflammatory variants, infiltration of neutrophils with variable numbers of eosinophils, monocytes, and lymphocytes are found in the upper dermis. In the classical mechanobullous variant, fibrosis and milia may be present which correspond to cicatricial changes seen clinically [Citation11]. Histopathology alone cannot differentiate between the different pemphigoid disorders.
6.2. Immunoelectron microscopy
Using electron microscopy ultrastructural changes in the perilesional skin, such as paucity or absence of anchoring fibrils can be demonstrated. This finding is similar to that of epidermolysis bullosa hereditaria and could explain skin fragility [Citation111,Citation112]. When performed from lesional skin, the cleavage plane can be demonstrated. However, cleavage can appear in sublamina densa or lamina lucida zone [Citation113]. Localization of blister in the lamina lucida has been explained by the lamina lucida being the locus minoris resistentiae and thus, most vulnerable to proteolytic enzymes released during the inflammatory reaction at the BMZ [Citation114–Citation116]. Thus, transmission electron microscopy cannot be used in differentiating EBA from inherited epidermolysis bullosa and other pemphigoid disorders.
In contrast, direct immunogold electron microscopy allows to visualize the deposits of autoantibodies in the sublamina densa which clearly differentiates EBA from deposits in other pemphigoid diseases located either in the lamina lucida or lamina densa [Citation117]. Subsequently, direct immunogold electron microscopy became the diagnostic gold standard for EBA [Citation85,Citation108,Citation118–Citation121]. Unfortunately, this technique is available for the diagnosis of autoimmune blistering diseases in only a handful of laboratories. Although attempts have been made that allow the sending of biopsies for direct immunogold electron microscopy [Citation122], this approach is best performed with freshly taken biopsies.
6.3. Direct IF microscopy
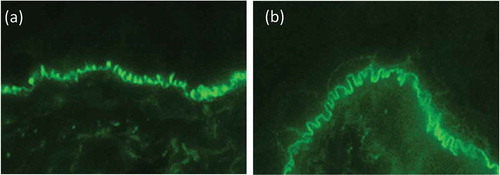
Direct IF microscopy is performed in a perilesional biopsy, i.e. adjacent (with a radius of 1 cm) to a macroscopic blister [Citation123]. In a lesional biopsy, i.e. when a split formation can be seen microscopically, direct IF microscopy can be false negative due to the proteolytic degradation of Ig deposits at the BMZ or false positive due to unspecific binding if autoantibodies at the BMZ of the microscopic splitting [Citation123]. In EBA, like in all pemphigoid diseases, linear binding of IgG and/or IgA, and/or C3 can be seen along the dermal–epidermal junction. Importantly, by close inspection of thin sections of 4–6 µm, Vodegel et al. could show that IgG/IgA linear deposits at the BMZ show a subtle but well remarkable pattern described as u-serrated [Citation110]. This pattern is characterized by arches that are closed at the bottom giving the appearance of ‘growing grass’ or ‘upstanding arms’ (). It is recommended to perform this analysis at ×400–×600 magnification. The u-serrated pattern is pathognomonic for skin-bound autoantibodies against type VII collagen as found in EBA and bullous systemic lupus erythematosus [Citation26,Citation124]. In all other pemphigoid diseases, a n-serrated pattern characterized by arches closed at the top giving a ‘snake-like’ appearance [Citation110,Citation124,Citation125]. However, in mucosal biopsies and a number of skin samples, no u- or n-serrated pattern can be seen. Current studies are aimed at defining the best conditions for serration pattern analysis, determine its sensitivity and establish an automated serration pattern analysis.
Figure 6. Direct immunofluorescence microscopy. (a) u-serrated deposition of IgG along the basement membrane zone in a patient with EBA. In contrast, in all of the pemphigoid diseases, an n-serrated pattern is seen, as exemplified in a patient with bullous pemphigoid (b). Magnification x 1000.
In samples where no serration pattern can be identified, the perilesional biopsy can be subjected to incubation with 1 M NaCl solution which leads to cleavage within the lamina lucida [Citation126]. In EBA, autoantibodies and complement depositions can then be detected along the floor of the artificial split. This technique, however, is tricky and may result in the complete destruction of the skin sample. Moreover, patients with anti-p200/laminin γ1 pemphigoid and anti-laminin 332 pemphigoid also reveal autoantibodies bound at the blister floor and cannot be differentiated from EBA by this technique [Citation4,Citation127].
When the perilesional skin biopsy is subjected to co-incubation with antibodies against e.g. BP180, laminin 332, and type VII collagen, the fluorescence overlay antigen mapping (FOAM) technique allows the detection of co-localization of autoantibodies by overlay of the different pictures [Citation128–Citation130]. In EBA, labeling of patient’s tissue-bound autoantibodies e.g. with a green fluorescence dye and staining of the biopsy with an antihuman type VII collagen antibody labeled e.g. with a red fluorescence dye will show a yellowish staining along the BMZ by direct IF microscopy. This technology is only available in few laboratories worldwide.
6.4. Serology
Serology has become the major diagnostic approach in autoimmune blistering diseases including EBA [Citation131]. This development was fostered by the establishment of several enzyme-linked immunosorbent assay (ELISA) and indirect IF microscopy-based test systems that became widely available and are based on the use of the corresponding recombinant target antigen [Citation55,Citation65,Citation132–Citation136]. In EBA, serological diagnosis is hampered by the relatively low rate of circulating autoantibodies which in some studies, was only found in about 60% of patients [Citation26,Citation137].
6.4.1. Indirect IF microscopy
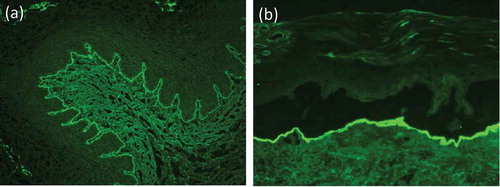
Indirect IF microscopy can be applied for the characterization of circulating antibodies. Standard substrates for this technique are monkey or rabbit esophagus as well as normal human skin in which subepidermal splitting was induced by incubation with 1 M NaCl solution () [Citation3,Citation123,Citation131]. As in all pemphigoid diseases, antitype VII collagen antibodies bind along the BMZ on the monkey and rabbit esophagus. By indirect IF microscopy of human salt-split skin, antitype VII collagen antibodies label the floor of the artificial blister () [Citation138]. However, antibodies against laminin 332 and the p200 protein/laminin γ1 also bind to the floor of the blister [Citation3,Citation127,Citation139,Citation140]. In latter two diseases, however, direct IF microscopy reveals an n-serrated pattern. Several studies have reported sensitivities of human salt-split skin for the detection of serum autoantibodies in EBA ranging from 57% to 100% with a mean of 64% in 191 EBA sera [Citation26,Citation27,Citation137,Citation141–Citation143].
Figure 7. Indirect immunofluorescence microscopy. (a) Linear binding of IgG along the basement membrane on monkey esophagus. (b) IgG locates to the floor of the artificial blister in 1 M NaCl-split human skin.
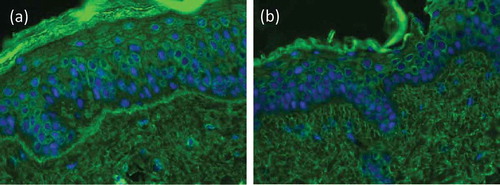
An elegant and relatively simple method for the detection of antitype VII collagen antibodies by indirect IF microscopy is the use of type VII collagen-deficient skin from patient with dystrophic epidermolysis bullosa. Incubation with EBA sera with type VII collagen-specific antibodies will not show any labeling of the BMZ, whereas on normal/salt-split human skin, as well as on laminin 332 and type XVII collagen-deficient skin, linear fluorescence is seen at the BMZ () [Citation144,Citation145].
Figure 8. Indirect immunofluorescence on type VII collagen-deficient skin. Indirect immunofluorescence microscopy of a EBA patient’s serum on normal human skin shows linear labeling of IgG at the basement membrane zone (a), while on type VII collagen-deficient skin, no staining is seen (b; courtesy of Dr. Hendri Pas, Department of Dermatology, University of Groningen, The Netherlands).
6.4.2. Western blotting
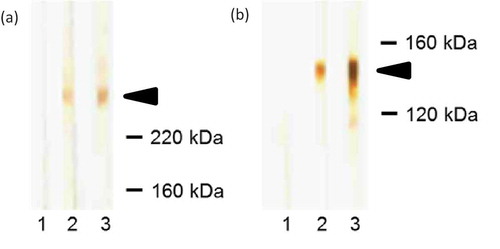
This semiquantitative method is based on cell-derived or recombinant forms of type VII collagen that had been subjected to SDS–PAGE, transferred onto a nitrocellulose membrane, and incubated with patient’s serum. Antitype VII collagen antibodies bound to the nitrocellulose-attached antigen can then be visualized. Extract of human dermis and the recombinant NC1 domain of type VII collagen are usually used [Citation52,Citation53,Citation146]. In dermal extract, antitype VII collagen antibodies bind to a 290-kDa protein which represents full-length type VII collagen () [Citation52,Citation53]. Moreover, type VII collagen has also been found in extracts of cultured human keratinocytes and human amniotic membrane that consequently, were proposed as useful in the detection of serum antitype VII collagen antibodies by immunoblotting [Citation147–Citation149].
Figure 9. Western blotting. (a) Western blot with dermal extract. 1, blood donor; 2, positive control; 3, EBA patient; arrow indicates the migration position of type VII collagen. (b) Western blot with recombinant NC1 domain of type VII collagen. 1, blood donor; 2, positive control; 3, EBA patient; arrow indicates the migration position of NC1 domain.
6.4.3. Enzyme-linked immunosorbent assay
In 1997, Chen and coworkers developed an ELISA based on the recombinant NC1 domain of type VII collagen with a sensitivity of 100% demonstrating that the NC1 domain was the immunodominant stretch of type VII collagen [Citation109]. This ELISA, however, was only available in this particular laboratory. In 2011 and 2013, two ELISA systems became widely available based on the combination of the recombinant NC1 and NC2 domains and the NC1 domain alone, respectively (MBL, Nagoya, Japan; Euroimmun, Lübeck, Germany) [Citation55,Citation65]. These assays revealed sensitivities of 93.8% and 94.5% and specificities of 98.1% and 98.7%, respectively, in large cohorts of 49 and 73 EBA sera [Citation55,Citation65]. Sera were, however, selected based on the clinical picture, and the presence of circulating autoantibodies by indirect IF microscopy on salt-split human skin and/or immunoblotting with cell-derived type VII collagen. When these ELISA were probed with EBA sera diagnosed by serration pattern analysis of direct IF microscopy or direct immunogold electron microscopy, much lower sensitivities of 53% and 30% are reported [Citation137,Citation150]. In EBA sera unreactive by indirect IF microscopy on human salt-split skin, ELISA reactivity could only be detected in 12% and 23% [Citation137,Citation150].
Most recently, two multivariant ELISA systems became widely available (Euroimmun, MBL). These assays allow the simultaneous testing of sera for IgG autoantibodies against the most relevant target antigens of autoimmune blistering diseases including type VII collagen [Citation151] (van Beek et al., submitted).
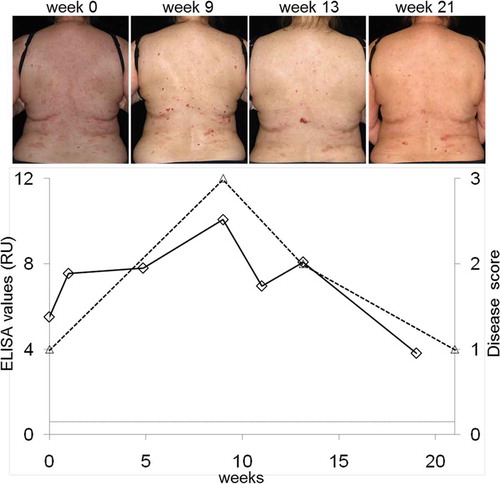
Compared to Western blotting, ELISA is significantly faster, more convenient, and better standardized. Furthermore, ELISA allows quantitative measurement of antitype VII collagen IgG and may, thus, be a valuable tool in the follow-up of EBA patients during the course of their disease. In fact, ELISA values were shown to correlate with disease activity in EBA patients () [Citation150,Citation152]. Recently, an in-house ELISA with a recombinant form of full-length type VII collagen was reported with a sensitivity of 65% [Citation137].
Figure 10. Correlation of serum levels of IgG antibodies against type VII collagen with disease severity. Serum autoantibody levels were detected by ELISA (diamonds; Euroimmun, Lübeck, Germany), disease activity was measured by a clinical score (triangles; 3, >10 lesions; 2, 4–10 lesions; 1, 1–3 lesions; 0, no lesions). Representative clinical pictures during the course of the disease are shown at the top.
6.4.4. BIOCHIP® mosaic
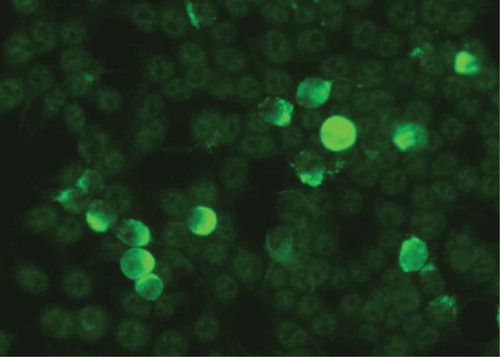
Indirect IF microscopy based on BIOCHIP® mosaics allows the simultaneous detection of serum autoantibodies against various substrates in a miniature incubation field of only several millimeters within a normal laboratory slide (Euroimmun). In autoimmune blistering diseases, a BIOCHIP® mosaic containing six substrates comprising monkey esophagus, salt-split skin, recombinant BP180 NC16A, and HEK293 cells transfected for expression of desmoglein 1, desmoglein 3, and BP230 is widely used in the dermatological community [Citation136,Citation153–Citation156]. Subsequently, this BIOCHIP® mosaic has been extended by HEK293 cells expressing the recombinant NC1 domain of type VII collagen on their cell surface revealing a sensitivity and specificity of 91.8% and 99.8% () [Citation65]. Other investigators also showed sensitivities of the type VII collagen-specific BIOCHIP® comparable with the corresponding ELISA [Citation137,Citation157]. As pointed out above, the sensitivity of detection assays for antitype VII collagen IgG may vary considerably depending on the analyzed patient cohort with circulating autoantibodies reported to be only present in about 60% EBA patients [Citation26].
Figure 11. Indirect immunofluorescence on HEK 293 cells expressing the recombinant NC1 domain of type VII collagen on their cell surface.
6.5. Diagnostic pathway
In a patient with a clinical picture compatible with EBA, the current diagnostic gold standard is the direct IF microscopy of a perilesional skin biopsy including serration pattern analysis. When an u-serrated pattern is observed EBA can be diagnosed. Direct immunogold electron microscopy may have a similar sensitivity, however, is a sophisticated procedure only available in few centers for the diagnosis of EBA. Even when diagnosis of EBA has been established by pattern analysis of direct IF or immunogold electron microscopy serum should be analyzed for autoantibodies against type VII collagen.
When the serration pattern cannot be determined serological analysis is favored. By indirect IF microscopy on human salt-split skin, EBA sera bind to the dermal side of the artificial split. However, this method is not diagnostic since sera from patients with anti-laminin 332 and anti-p200/laminin γ1 pemphigoid also label the floor of the artificial split. Serological diagnosis of EBA requires the detection of antitype VII collagen autoantibodies. At present, the most convenient and standardized assays are widely available ELISA systems and an indirect IF test based on the use of recombinant fragments of type VII collagen (Euroimmun, MBL) [Citation55,Citation65]. In addition, several in-house systems are in use such as immunoblotting with various cell-derived or recombinant forms of type VII collagen. These assays are less standardized but have a role e.g. in detecting IgA antibodies against type VII collagen which cannot be achieved by the commercial assays.
A highly reliable, however, also not widely available approach, is the use of type VII collagen-deficient human skin as substrate for indirect IF microscopy. If no circulating antitype VII collagen antibodies are detectable and an undetermined serration pattern is seen by direct IF microscopy, FOAM may be applied [Citation4]. The suggested diagnostic procedure in EBA is schematically shown in .
Figure 12. Diagnostic pathway for EBA.
1even when the diagnosis of EBA can be made based on an u-serrated pattern, detection of serum anti-type VII collagen antibodies is recommended; 2commercially available; 3depending of availability; positivity in any of the 4 assays will allow diagnosis of EBA; 4only available in specialized laboratories; 5from patients with dystrophic epidermolysis bullosa; 6bullous pemphigoid (BP), predominant IgG reactivity by direct and/or indirect IF microscopy, no floor binding by indirect IF microscopy, and no predominant mucosal involvement; mucous membrane pemphigoid (MMP), predominant mucosal involvement, when floor binding by indirect IF microscopy, laminin 332 reactivity needs to be analyzed; linear IgA diseases (LAD), predominant IgA reactivity by direct and/or indirect IF microscopy; anti-p200 pemphigoid, reactivity with the p200 protein and/or laminin γ1 [Citation4].
![Figure 12. Diagnostic pathway for EBA.1even when the diagnosis of EBA can be made based on an u-serrated pattern, detection of serum anti-type VII collagen antibodies is recommended; 2commercially available; 3depending of availability; positivity in any of the 4 assays will allow diagnosis of EBA; 4only available in specialized laboratories; 5from patients with dystrophic epidermolysis bullosa; 6bullous pemphigoid (BP), predominant IgG reactivity by direct and/or indirect IF microscopy, no floor binding by indirect IF microscopy, and no predominant mucosal involvement; mucous membrane pemphigoid (MMP), predominant mucosal involvement, when floor binding by indirect IF microscopy, laminin 332 reactivity needs to be analyzed; linear IgA diseases (LAD), predominant IgA reactivity by direct and/or indirect IF microscopy; anti-p200 pemphigoid, reactivity with the p200 protein and/or laminin γ1 [Citation4].](/cms/asset/04c2a00e-6ab4-4c9f-952f-eeda60049b08/ierm_a_1221343_f0012_oc.jpg)
7. Expert commentary
Despite recent progress in our understanding of the pathophysiology of EBA and development of novel diagnostic methods, diagnosis and management of EBA still remain challenging. This is due to the low incidence of EBA and the related lack of prospective controlled clinical trials. Diagnosis is complicated by the polymorphous clinical picture and the sophisticated diagnostic approach summarized in . Most EBA patients are diagnosed and treated in specialized centers experienced in the management of AIBDs.
8. Five-year view
Considerable progress has been made in the diagnosis of EBA by the introduction of serration pattern diagnosis of direct IF microscopy and the availability of commercial test systems for the detection of serum IgG against type VII collagen. While the serological tests are already widely used in the community, the concept of serration pattern analysis needs to be further promoted and underlined by additional studies that are currently being performed. An international guideline of the diagnosis of EBA is being discussed in the community for some time and will be finalized within the next 1 or 2 years. Due to the availability of robust mouse models of EBA, valuable data about the pathophysiology of pemphigoid diseases and autoantibody-mediated diseases will be generated that may also help to better understand the human disorders including EBA. Moreover, the use of the EBA mouse models in preclinical trials will bring to life novel therapeutic strategies and anti-inflammatory mediators that will hopefully also relieve the considerable and so far unmet health burden of patients with EBA.
9. Key issues
EBA is a rare chronic autoimmune blistering disease of skin and mucous membranes caused by antibodies against type VII collagen
Clinical manifestations of EBA are highly variable with several well defined phenotypes, i.e. classical mechanobullous from and four inflammatory variants resembling bullous pemphigoid, mucous membrane pemphigoid, linear IgA-like disease, and Brunsting-Perry pemphigoid
The current diagnostic gold standard is the direct IF microscopy of a perilesional skin biopsy. Linear deposits of IgG, IgA and/or C3 is seen along the BMZ. An u-serrated pattern is diagnostic for EBA or bullous systemic lupus erythematosus.
Detection of serum autoantibodies against type VII collagen is also diagnostic. Several test systems for the serological diagnosis of EBA have recently been introduced in the market.
Since serum autoantibodies against type VII collagen can only be detected in about 60% of EBA patients, specialized laboratories that offer a panel of additional sophisticated approaches for the diagnosis of EBA are required.
Declaration of interest
E Schmidt has a scientific cooperation with Euroimmun. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Acknowledgements
We are grateful to Vanessa Krull for performing the immunoblots shown in .
Additional information
Funding
References
- Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347(12):911–920.
- Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol. 2016;11:175–197.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381(9863):320–332.
- Schmidt E, Groves R. Immunobullous diseases. In: Griffith C, Barker J, Chalmers R, Bleiker T, Creamer D, editors. Rook’s textbook of dermatology, part 3, chapter 50. 9th ed. Chichester: Wiley-Blackwell; 2016. Vol. 50, p. 1–56.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;812029:2013.
- Kasperkiewicz M, Sadik CD, Bieber K, et al. Epidermolysis bullosa acquisita: from pathophysiology to novel therapeutic options. J Invest Dermatol. 2016;136(1):24–33.
- Keene DR, Sakai LY, Lunstrum GP, et al. Type VII collagen forms an extended network of anchoring fibrils. J Cell Biol. 1987;104(3):611–621.
- Lunstrum GP, Sakai LY, Keene DR, et al. Large complex globular domains of type VII procollagen contribute to the structure of anchoring fibrils. J Biol Chem. 1986;261(19):9042–9048.
- Sakai LY, Keene DR, Morris NP, et al. Type VII collagen is a major structural component of anchoring fibrils. J Cell Biol. 1986;103(4):1577–1586.
- Roenigk HH Jr, Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita. Report of three cases and review of all published cases. Arch Dermatol. 1971;103(1):1–10.
- Gammon WR, Briggaman RA, Woodley DT, et al. Epidermolysis bullosa acquisita–a pemphigoid-like disease. J Am Acad Dermatol. 1984;11(5 Pt 1):820–832.
- Kurzhals G, Stolz W, Meurer M, et al. Acquired epidermolysis bullosa with the clinical feature of Brunsting-Perry cicatricial bullous pemphigoid. Arch Dermatol. 1991;127(3):391–395.
- Zambruno G, Manca V, Kanitakis J, et al. Linear IgA bullous dermatosis with autoantibodies to a 290 kd antigen of anchoring fibrils. J Am Acad Dermatol. 1994;31(5 Pt 2):884–888.
- Dahl MG. Epidermolysis bullosa acquisita–a sign of cicatricial pemphigoid? Br J Dermatol. 1979;101(4):475–484.
- Elliott GT. Two cases of epidermolysis bullosa. J Cutan Genitourin Dis. 1895;13:10–18.
- Woodley DT, Briggaman RA, O’Keefe EJ, et al. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310(16):1007–1013.
- Woodley DT, Burgeson RE, Lunstrum G, et al. Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of type VII procollagen. J Clin Invest. 1988;81(3):683–687.
- Borradori L, Sonnenberg A. Structure and function of hemidesmosomes: more than simple adhesion complexes. J Invest Dermatol. 1999;112(4):411–418.
- Bernard P, Vaillant L, Labeille B, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol. 1995;131(1):48–52.
- Bertram F, Brocker EB, Zillikens D, et al. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges. 2009;7(5):434–440.
- Zillikens D, Wever S, Roth A, et al. Incidence of autoimmune subepidermal blistering dermatoses in a region of central Germany. Arch Dermatol. 1995;131(8):957–958.
- Nanda A, Dvorak R, Al-Saeed K, et al. Spectrum of autoimmune bullous diseases in Kuwait. Int J Dermatol. 2004;43(12):876–881.
- Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol. 2002;147(3):476–480.
- Hübner F, Recke A, Zillikens D, et al. Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Invest Dermatol. 2016. doi:10.1016/j.jid.2016.07.013.
- Zumelzu C, Le Roux-Villet C, Loiseau P, et al. Black patients of African descent and HLA-DRB115:03 frequency overrepresented in epidermolysis bullosa acquisita. J Invest Dermatol. 2011;131(12):2386–2393.
- Buijsrogge JJ, Diercks GF, Pas HH, et al. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol. 2011;165(1):92–98.
- Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol. 2011;91(3):307–312.
- Gammon WR. Epidermolysis bullosa acquisita. Semin Dermatol. 1988;7(3):218–224.
- Arpey CJ, Elewski BE, Moritz DK, et al. Childhood epidermolysis bullosa acquisita. Report of three cases and review of literature. J Am Acad Dermatol. 1991;24((5 Pt 1):706–714.
- Guerra L, Pacifico V, Calabresi V, et al. Childhood epidermolysis bullosa acquisita during squaric acid dibutylester (SADBE) immunotherapy for alopecia areata. Br J Dermatol. 2016. doi:10.1111/bjd.14764
- Gammon WR, Briggaman RA. Epidermolysis bullosa acquisita and bullous systemic lupus erythematosus. Diseases of autoimmunity to type VII collagen. Dermatol Clin. 1993;11(3):535–547.
- Gammon WR, Heise ER, Burke WA, et al. Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa acquisita antigen: evidence that the expression of autoimmunity to type VII collagen is HLA class II allele associated. J Invest Dermatol. 1988;91(3):228–232.
- Lee CW, Kim SC, Han H. Distribution of HLA class II alleles in Korean patients with epidermolysis bullosa acquisita. Dermatology. 1996;193(4):328–329.
- Ludwig RJ, Recke A, Bieber K, et al. Generation of antibodies of distinct subclasses and specificity is linked to H2s in an active mouse model of epidermolysis bullosa acquisita. J Invest Dermatol. 2011;131(1):167–176.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118(6):1059–1064.
- Hundorfean G, Neurath MF, Sitaru C. Autoimmunity against type VII collagen in inflammatory bowel disease. J Cell Mol Med. 2010;14(10):2393–2403.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37(3):220–230.
- Chen M, Kim GH, Prakash L, et al. Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45(1):91–101.
- Schulze F, Neumann K, Recke A, et al. Malignancies in pemphigus and pemphigoid diseases. J Invest Dermatol. 2015;135(5):1445–1447.
- Aractingi S, Bachmeyer C, Prost C, et al. Subepidermal autoimmune bullous skin diseases associated with B-cell lymphoproliferative disorders. Medicine (Baltimore). 1999;78(4):228–235.
- Delbaldo C, Chen M, Friedli A, et al. Drug-induced epidermolysis bullosa acquisita with antibodies to type VII collagen. J Am Acad Dermatol. 2002;46(5 Suppl):S161–164.
- Jappe U, Zillikens D, Bonnekoh B, et al. Epidermolysis bullosa acquisita with ultraviolet radiationsensitivity. Br J Dermatol. 2000;142(3):517–520.
- Baican A, Chiriac G, Baican C, et al. Metal sensitization precipitates skin blistering in epidermolysis bullosa acquisita. J Dermatol. 2010;37(3):280–282.
- Wakelin SH, Allen J, Zhou S, et al. Drug-induced linear IgA disease with antibodies to collagen VII. Br J Dermatol. 1998;138(2):310–314.
- Burgeson RE. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J Invest Dermatol. 1993;101(3):252–255.
- Chen M, Marinkovich MP, Jones JC, et al. NC1 domain of type VII collagen binds to the beta3 chain of laminin 5 via a unique subdomain within the fibronectin-like repeats. J Invest Dermatol. 1999;112(2):177–183.
- Chen M, Marinkovich MP, Veis A, et al. Interactions of the amino-terminal noncollagenous (NC1) domain of type VII collagen with extracellular matrix components. A potential role in epidermal-dermal adherence in human skin. J Biol Chem. 1997;272(23):14516–14522.
- Rousselle P, Keene DR, Ruggiero F, et al. Laminin 5 binds the NC-1 domain of type VII collagen. J Cell Biol. 1997;138(3):719–728.
- Brittingham R, Uitto J, Fertala A. High-affinity binding of the NC1 domain of collagen VII to laminin 5 and collagen IV. Biochem Biophys Res Commun. 2006;343(3):692–699.
- Villone D, Fritsch A, Koch M, et al. Supramolecular interactions in the dermo-epidermal junction zone: anchoring fibril-collagen VII tightly binds to banded collagen fibrils. J Biol Chem. 2008;283(36):24506–24513.
- Woodley DT, O’Keefe EJ, McDonald JA, et al. Specific affinity between fibronectin and the epidermolysis bullosa acquisita antigen. J Clin Invest. 1987;79(6):1826–1830.
- Jones DA, Hunt SW III, Prisayanh PS, et al. Immunodominant autoepitopes of type VII collagen are short, paired peptide sequences within the fibronectin type III homology region of the noncollagenous (NC1) domain. J Invest Dermatol. 1995;104(2):231–235.
- Lapiere JC, Woodley DT, Parente MG, et al. Epitope mapping of type VII collagen. Identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. J Clin Invest. 1993;92(4):1831–1839.
- Ishii N, Yoshida M, Ishida-Yamamoto A, et al. Some epidermolysis bullosa acquisita sera react with epitopes within the triple-helical collagenous domain as indicated by immunoelectron microscopy. Br J Dermatol. 2009;160(5):1090–1093.
- Saleh MA, Ishii K, Kim YJ, et al. Development of NC1 and NC2 domains of type VII collagen ELISA for the diagnosis and analysis of the time course of epidermolysis bullosa acquisita patients. J Dermatol Sci. 2011;62(3):169–175.
- Callot-Mellot C, Bodemer C, Caux F, et al. Epidermolysis bullosa acquisita in childhood. Arch Dermatol. 1997;133(9):1122–1126.
- Tanaka H, Ishida-Yamamoto A, Hashimoto T, et al. A novel variant of acquired epidermolysis bullosa with autoantibodies against the central triple-helical domain of type VII collagen. Lab Invest. 1997;77(6):623–632.
- Park SB, Cho KH, Youn JL, et al. Epidermolysis bullosa acquisita in childhood–a case mimicking chronic bullous dermatosis of childhood. Clin Exp Dermatol. 1997;22(5):220–222.
- Schmidt E, Hopfner B, Chen M, et al. Childhood epidermolysis bullosa acquisita: a novel variant with reactivity to all three structural domains of type VII collagen. Br J Dermatol. 2002;147(3):592–597.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30(1):60–69.
- Vorobyev A, Ujiie H, Recke A, et al. Autoantibodies to multiple epitopes on the non-collagenous-1 domain of Type VII collagen induce blisters. J Invest Dermatol. 2015;135(6):1565–1573.
- Chen M, Keene DR, Costa FK, et al. The carboxyl terminus of type VII collagen mediates antiparallel dimer formation and constitutes a new antigenic epitope for epidermolysis Bullosa acquisita autoantibodies. J Biol Chem. 2001;276(24):21649–21655.
- Bernard P, Prost C, Aucouturier P, et al. The subclass distribution of IgG autoantibodies in cicatricial pemphigoid and epidermolysis bullosa acquisita. J Invest Dermatol. 1991;97(2):259–263.
- Oostingh GJ, Sitaru C, Zillikens D, et al. Subclass distribution of type VII collagen-specific autoantibodies in patients with inflammatory bowel disease. J Dermatol Sci. 2005;37(3):182–184.
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2012;68(3):e89–95.
- Gandhi K, Chen M, Aasi S, et al. Autoantibodies to type VII collagen have heterogeneous subclass and light chain compositions and their complement-activating capacities do not correlate with the inflammatory clinical phenotype. J Clin Immunol. 2000;20(6):416–423.
- Sitaru C, Mihai S, Otto C, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest. 2005;115(4):870–878.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177(5):3461–3468.
- Sitaru C, Kromminga A, Hashimoto T, et al. Autoantibodies to type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am J Pathol. 2002;161(1):301–311.
- Borradori L, Caldwell JB, Briggaman RA, et al. Passive transfer of autoantibodies from a patient with mutilating epidermolysis bullosa acquisita induces specific alterations in the skin of neonatal mice. Arch Dermatol. 1995;131(5):590–595.
- Chen M, Doostan A, Bandyopadhyay P, et al. The cartilage matrix protein subdomain of type VII collagen is pathogenic for epidermolysis bullosa acquisita. Am J Pathol. 2007;170(6):2009–2018.
- Woodley DT, Chang C, Saadat P, et al. Evidence that anti-type VII collagen antibodies are pathogenic and responsible for the clinical, histological, and immunological features of epidermolysis bullosa acquisita. J Invest Dermatol. 2005;124(5):958–964.
- Umemoto H, Akiyama M, Domon T, et al. Type VII collagen deficiency causes defective tooth enamel formation due to poor differentiation of ameloblasts. Am J Pathol. 2012;181(5):1659–1671
- Iwata H, Bieber K, Tiburzy B, et al. B cells, dendritic cells, and macrophages are required to induce an autoreactive CD4 helper T cell response in experimental epidermolysis bullosa acquisita. J Immunol. 2013;191(6):2978–2988.
- Hellberg L, Samavedam UK, Holdorf K, et al. Methylprednisolone blocks autoantibody-induced tissue damage in experimental models of bullous pemphigoid and epidermolysis bullosa acquisita through inhibition of neutrophil activation. J Invest Dermatol. 2013;133(10):2390–2399.
- Sadeghi H, Gupta Y, Moller S, et al. The retinoid-related orphan receptor alpha is essential for the end-stage effector phase of experimental epidermolysis bullosa acquisita. J Pathol. 2015;237(1):111–122.
- Tukaj S, Hellberg L, Ueck C, et al. Heat shock protein 90 is required for ex vivo neutrophil-driven autoantibody-induced tissue damage in experimental epidermolysis bullosa acquisita. Exp Dermatol. 2015;24(6):471–473.
- Kasperkiewicz M, Muller R, Manz R, et al. Heat-shock protein 90 inhibition in autoimmunity to type VII collagen: evidence that nonmalignant plasma cells are not therapeutic targets. Blood. 2011;117(23):6135–6142.
- Mihai S, Chiriac MT, Takahashi K, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol. 2007;178(10):6514–6521.
- Chiriac MT, Roesler J, Sindrilaru A, et al. NADPH oxidase is required for neutrophil-dependent autoantibody-induced tissue damage. J Pathol. 2007;212(1):56–65.
- Ellebrecht CT, Srinivas G, Bieber K, et al. Skin microbiota-associated inflammation precedes autoantibody induced tissue damage in experimental epidermolysis bullosa acquisita. J Autoimmun. 2016;68:14–22.
- Hirose M, Vafia K, Kalies K, et al. Enzymatic autoantibody glycan hydrolysis alleviates autoimmunity against type VII collagen. J Autoimmun. 2012;39(4):304–314.
- Karsten CM, Pandey MK, Figge J, et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med. 2012;18(9):1401–1406.
- Sadeghi H, Lockmann A, Hund AC, et al. Caspase-1-independent IL-1 release mediates blister formation in autoantibody-induced tissue injury through modulation of endothelial adhesion molecules. J Immunol. 2015;194(8):3656–3663.
- Briggaman RA, Gammon WR, Woodley DT. Epidermolysis bullosa acquisita of the immunopathological type (dermolytic pemphigoid). J Invest Dermatol. 1985;85(1 Suppl):79s–84s.
- Joly P, Baricault S, Sparsa A, et al. Incidence and mortality of bullous pemphigoid in France. J Invest Dermatol. 2012;132(8):1998–2004.
- Langan SM, Smeeth L, Hubbard R, et al. Bullous pemphigoid and pemphigus vulgaris–incidence and mortality in the UK: population based cohort study. Bmj. 2008;337:a180.
- Marazza G, Pham HC, Scharer L, et al. Incidence of bullous pemphigoid and pemphigus in Switzerland: a 2-year prospective study. Br J Dermatol. 2009;161(4):861–868.
- Schmidt E, Borradori L, Joly P. Epidemiology of autoimmune bullous diseases. In: Murrell D, editors. Blistering diseases. 1st ed. Heidelberg: Springer; 2015. p. 251–264.
- Schmidt E, Della Torre R, Borradori L. Clinical features and practical diagnosis of bullous pemphigoid. Dermatol Clin. 2011;29(3):427–438, viii–ix.
- Gammon WR, Briggaman RA, Wheeler CE Jr Epidermolysis bullosa acquisita presenting as an inflammatory bullous disease. J Am Acad Dermatol. 1982;7(3):382–387.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138(3):370–379.
- Luke MC, Darling TN, Hsu R, et al. Mucosal morbidity in patients with epidermolysis bullosa acquisita. Arch Dermatol. 1999;135(8):954–959.
- Letko E, Bhol K, Anzaar F, et al. Chronic cicatrizing conjunctivitis in a patient with epidermolysis bullosa acquisita. Arch Ophthalmol. 2006;124(11):1615–1618.
- Lang PG Jr, Tapert MJ. Severe ocular involvement in a patient with epidermolysis bullosa acquisita. J Am Acad Dermatol. 1987;16(2 Pt 2):439–443.
- Woodley DT, Briggaman RA, Gammon WT. Review and update of epidermolysis bullosa acquisita. Semin Dermatol. 1988;7(2):111–122.
- Alexandre M, Brette MD, Pascal F, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (Baltimore). 2006;85(4):239–252.
- Schattenkirchner S, Lemann M, Prost C, et al. Localized epidermolysis bullosa acquisita of the esophagus in a patient with Crohn’s disease. Am J Gastroenterol. 1996;91(8):1657–1659.
- Vodegel RM, De Jong MC, Pas HH, et al. IgA-mediated epidermolysis bullosa acquisita: two cases and review of the literature. J Am Acad Dermatol. 2002;47(6):919–925.
- Bauer JW, Schaeppi H, Metze D, et al. Ocular involvement in IgA-epidermolysis bullosa acquisita. Br J Dermatol. 1999;141(5):887–892.
- Hashimoto T, Ishiko A, Shimizu H, et al. A case of linear IgA bullous dermatosis with IgA anti-type VII collagen autoantibodies. Br J Dermatol. 1996;134(2):336–339.
- Ls Cm C, Woodley DT. Epidermolysis bullosa acquisita in the elderly. Clinical manifestationsdiagnosis and therapy. J Geriatr Dermatol. 1996;4:47–52.
- Brunsting LA, Perry HO. Benign pemphigold; a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm. 1957;75(4):489–501.
- Lee CW, Jun KM. Epidermolysis bullosa acquisita presenting with localized facial blisters. Clin Exp Dermatol. 1992;17(5):363–365.
- Choi GS, Lee ES, Kim SC, et al. Epidermolysis bullosa acquisita localized to the face. J Dermatol. 1998;25(1):19–22.
- Joly P, Ruto F, Thomine E, et al. Brunsting-Perry cicatricial bullous pemphigoid: a clinical variant of localized acquired epidermolysis bullosa? J Am Acad Dermatol. 1993;28(1):89–92.
- Abrams ML, Smidt A, Benjamin L, et al. Congenital epidermolysis bullosa acquisita: vertical transfer of maternal autoantibody from mother to infant. Arch Dermatol. 2010;147(3):337–341.
- Yaoita H, Briggaman RA, Lawley TJ, et al. Epidermolysis bullosa acquisita: ultrastructural and immunological studies. J Invest Dermatol. 1981;76(4):288–292.
- Chen M, Chan LS, Cai X, et al. Development of an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis bullosa acquisita. J Invest Dermatol. 1997;108(1):68–72.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151(1):112–118.
- Woodley DT, Remington J, Chen M. Autoimmunity to type VII collagen: epidermolysis bullosa acquisita. Clin Rev Allergy Immunol. 2007;33(1–2):78–84.
- Gibbs RB, Minus HR. Epidermolysis bullosa acquisita with electron microscopical studies. Arch Dermatol. 1975;111(2):215–220.
- Fine JD, Tyring S, Gammon WR. The presence of intra-lamina lucida blister formation in epidermolysis bullosa acquisita: possible role of leukocytes. J Invest Dermatol. 1989;92(1):27–32.
- Briggaman RA, Schechter NM, Fraki J, et al. Degradation of the epidermal-dermal junction by proteolytic enzymes from human skin and human polymorphonuclear leukocytes. J Exp Med. 1984;160(4):1027–1042.
- Klein GF, Hintner H, Schuler G, et al. Junctional blisters in acquired bullous disorders of the dermal-epidermal junction zone: role of the lamina lucida as the mechanical locus minoris resistentiae. Br J Dermatol. 1983;109(5):499–508.
- Briggaman RA, Wheeler CE Jr The epidermal-dermal junction. J Invest Dermatol. 1975;65(1):71–84.
- Nieboer C, Boorsma DM, Woerdeman MJ, et al. Epidermolysis bullosa acquisita. Immunofluorescence, electron microscopic and immunoelectron microscopic studies in four patients. Br J Dermatol. 1980;102(4):383–392.
- Prost C, Labeille B, Chaussade V, et al. Immunoelectron microscopy in subepidermal autoimmune bullous diseases: a prospective study of IgG and C3 bound in vivo in 32 patients. J Invest Dermatol. 1987;89(6):567–573.
- Prost C, Dubertret L, Fosse M, et al. A routine immuno-electron microscopic technique for localizing an auto-antibody on epidermal basement membrane. Br J Dermatol. 1984;110(1):1–7.
- Shimizu H, McDonald JN, Gunner DB, et al. Epidermolysis bullosa acquisita antigen and the carboxy terminus of type VII collagen have a common immunolocalization to anchoring fibrils and lamina densa of basement membrane. Br J Dermatol. 1990;122(5):577–585.
- Karpati S, Stolz W, Meurer M, et al. In situ localization of IgG in epidermolysis bullosa acquisita by immunogold technique. J Am Acad Dermatol. 1992;26(5 Pt 1):726–730.
- Vaughn Jones SA, Palmer I, Bhogal BS, et al. The use of Michel’s transport medium for immunofluorescence and immunoelectron microscopy in autoimmune bullous diseases. J Cutan Pathol. 1995;22(4):365–370.
- Schmidt E, Goebeler M, Hertl M, et al. S2k guideline for the diagnosis of pemphigus vulgaris/foliaceus and bullous pemphigoid. J Dtsch Dermatol Ges. 2015;13(7):713–727.
- Terra JB, Meijer JM, Jonkman MF, et al. The n- vs. u-serration is a learnable criterion to differentiate pemphigoid from epidermolysis bullosa acquisita in direct immunofluorescence serration pattern analysis. Br J Dermatol. 2013;169(1):100–105.
- Terra JB, Pas HH, Hertl M, et al. Immunofluorescence serration pattern analysis as a diagnostic criterion in antilaminin-332 mucous membrane pemphigoid: immunopathological findings and clinical experience in 10 Dutch patients. Br J Dermatol. 2011;165(4):815–822.
- Woodley D, Sauder D, Talley MJ, et al. Localization of basement membrane components after dermal-epidermal junction separation. J Invest Dermatol. 1983;81(2):149–153.
- Goletz S, Hashimoto T, Zillikens D, et al. Anti-p200 pemphigoid. J Am Acad Dermatol. 2014;71(1):185–191.
- Bruins S, De Jong MC, Heeres K, et al. Fluorescence overlay antigen mapping of the epidermal basement membrane zone: III. Topographic staining and effective resolution. J Histochem Cytochem. 1995;43(7):649–656.
- De Jong MC, Bruins S, Heeres K, et al. Bullous pemphigoid and epidermolysis bullosa acquisita. Differentiation by fluorescence overlay antigen mapping. Arch Dermatol. 1996;132(2):151–157.
- Wozniak K, Kazama T, Kowalewski C. A practical technique for differentiation of subepidermal bullous diseases: localization of in vivo-bound IgG by laser scanning confocal microscopy. Arch Dermatol. 2003;139(8):1007–1011.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev. 2010;10(2):84–89.
- Blocker IM, Dahnrich C, Probst C, et al. Epitope mapping of BP230 leading to a novel enzyme-linked immunosorbent assay for autoantibodies in bullous pemphigoid. Br J Dermatol. 2012;166(5):964–970.
- Schmidt E, Dahnrich C, Rosemann A, et al. Novel ELISA systems for antibodies to desmoglein 1 and 3: correlation of disease activity with serum autoantibody levels in individual pemphigus patients. Exp Dermatol. 2010;19(5):458–463.
- Sitaru C, Dahnrich C, Probst C, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol. 2007;16(9):770–777.
- Van Beek N, Knuth-Rehr D, Altmeyer P, et al. Diagnostics of autoimmune bullous diseases in German dermatology departments. J Dtsch Dermatol Ges. 2012;10(7):492–499.
- Van Beek N, Rentzsch K, Probst C, et al. Serological diagnosis of autoimmune bullous skin diseases: Prospective comparison of the BIOCHIP mosaic-based indirect immunofluorescence technique with the conventional multi-step single test strategy. Orphanet J Rare Dis. 2012;7(1):49.
- Seta V, Aucouturier F, Bonnefoy J, et al. Comparison of 3 type VII collagen (C7) assays for serologic diagnosis of epidermolysis bullosa acquisita (EBA). J Am Acad Dermatol. 2016;74(6):1166–1172.
- Gammon WR, Briggaman RA, Inman AO III, et al. Differentiating anti-lamina lucida and anti-sublamina densa anti-BMZ antibodies by indirect immunofluorescence on 1.0 M sodium chloride-separated skin. J Invest Dermatol. 1984;82(2):139–144.
- Domloge-Hultsch N, Anhalt GJ, Gammon WR, et al. Antiepiligrin cicatricial pemphigoid. A subepithelial bullous disorder. Arch Dermatol. 1994;130(12):1521–1529.
- Zillikens D, Kawahara Y, Ishiko A, et al. A novel subepidermal blistering disease with autoantibodies to a 200-kDa antigen of the basement membrane zone. J Invest Dermatol. 1996;106(6):1333–1338.
- Calabresi V, Sinistro A, Cozzani E, et al. Sensitivity of different assays for the serological diagnosis of epidermolysis bullosa acquisita: analysis of a cohort of 24 Italian patients. J Eur Acad Dermatol Venereol. 2014;28(4):483–490.
- Delgado L, Aoki V, Santi C, et al. Clinical and immunopathological evaluation of epidermolysis bullosa acquisita. Clin Exp Dermatol. 2011;36(1):12–18.
- Ghohestani RF, Nicolas JF, Rousselle P, et al. Diagnostic value of indirect immunofluorescence on sodium chloride-split skin in differential diagnosis of subepidermal autoimmune bullous dermatoses. Arch Dermatol. 1997;133(9):1102–1107.
- Jonkman MF, Schuur J, Dijk F, et al. Inflammatory variant of epidermolysis bullosa acquisita with IgG autoantibodies against type VII collagen and laminin alpha3. Arch Dermatol. 2000;136(2):227–231.
- Vodegel RM, Kiss M, Cjm De Jong M, et al. The use of skin substrates deficient in basement membrane molecules for the diagnosis of subepidermal autoimmune bullous disease. Eur J Dermatol. 1998;8(2):83–85.
- Chen M, Costa FK, Lindvay CR, et al. The recombinant expression of full-length type VII collagen and characterization of molecular mechanisms underlying dystrophic epidermolysis bullosa. J Biol Chem. 2002;277(3):2118–2124.
- Grootenboer-Mignot S, Descamps V, Picard-Dahan C, et al. Place of human amniotic membrane immunoblotting in the diagnosis of autoimmune bullous dermatoses. Br J Dermatol. 2010;162(4):743–750.
- Oyama N, Bhogal BS, Carrington P, et al. Human placental amnion is a novel substrate for detecting autoantibodies in autoimmune bullous diseases by immunoblotting. Br J Dermatol. 2003;148(5):939–944.
- Batteux F, Franck N, Jaffray P, et al. An extract from cultured human keratinocytes that contains the major autoantigens related to autoimmune bullous skin diseases. J Clin Immunol. 1997;17(3):228–233.
- Terra JB, Jonkman MF, Diercks GF, et al. Low sensitivity of type VII collagen enzyme-linked immunosorbent assay in epidermolysis bullosa acquisita: serration pattern analysis on skin biopsy is required for diagnosis. Br J Dermatol. 2013;169(1):164–167.
- Horvath ON, Varga R, Kaneda M, et al. Diagnostic performance of the “MESACUP anti-Skin profile TEST”. Eur J Dermatol. 2016;26(1):56–63.
- Kim JH, Kim YH, Kim S, et al. Serum levels of anti-type VII collagen antibodies detected by enzyme-linked immunosorbent assay in patients with epidermolysis bullosa acquisita are correlated with the severity of skin lesions. J Eur Acad Dermatol Venereol. 2012;27(2):e224–230.
- Tampoia M, Zucano A, Villalta D, et al. Anti-skin specific autoantibodies detected by a new immunofluorescence multiplex biochip method in patients with autoimmune bullous diseases. Dermatology. 2012;225(1):37–44.
- Russo I, Saponeri A, Peserico A, et al. The use of biochip immunofluorescence microscopy for the diagnosis of Pemphigus vulgaris. Acta Histochem. 2014;116(5):713–716.
- Zarian H, Saponeri A, Michelotto A, et al. Biochip technology for the serological diagnosis of bullous pemphigoid. ISRN Dermatol. 2012;2012:237802.
- Gornowicz-Porowska J, Dmochowski M, Pietkiewicz P, et al. Mucosal-dominant pemphigus vulgaris in a captopril-taking woman with angioedema. An Bras Dermatol. 2015;90(5):748–751.
- Marzano AV, Cozzani E, Biasin M, et al. The use of Biochip immunofluorescence microscopy for the serological diagnosis of epidermolysis bullosa acquisita. Arch Dermatol Res. 2016;308(4):273–276.