
ABSTRACT
Introduction
There is a wide spectrum of noninfectious gastrointestinal pathology, causing considerable morbidity and mortality in CVID, where both etiology and effective therapy are under debate.
Areas covered
This review will focus on the noninfectious inflammation in the GI tract in CVID patients, covering the both the upper and lower GI tract inflammation, including the liver. The controversy of the CVID enteropathy definition and that of gluten-free diet for celiac-like disease in CVID will be discussed. Furthermore, the review will cover the link between GI inflammation and GI cancer. Finally, the role of gut microbiota, IgA, and genetics and its relationship with CVID enteropathy is scrutinized. The authors reviewed literature from PubMed.
Expert opinion
The heterogeneity and the unknown mechanism behind CVID enteropathy, and thereby the lack of effective treatment, is one of the key challenges in the field of CVID. Celiac-like disease in CVID is due to immune dysregulation, and a gluten-free diet is therefore not indicated. Gut microbial dysbiosis and mucosal IgA can initiate systemic and local inflammation and is involved in the immune dysregulation in CVID. Considering the heterogeneity of CVID enteropathy, personalized medicine is probably the future for these patients.
1. Introduction
Common variable immunodeficiency (CVID) is the most frequent symptomatic primary immunodeficiency in adulthood, affecting 1 in 25,000 to 1 in 50,000 Caucasians [Citation1]. These patients have a B-cell dysfunction that leads to hypogammaglobulinemia and is characterized by a marked decrease of Immunoglobulin (Ig)G and at least one of the two isotypes IgM or IgA. As a consequence, CVID patients have impaired antibody production to microbes and vaccines [Citation2]. Recurrent airway infections secondary to capsulated bacteria Streptococcus pneumonia, Haemophilus influenza, and Moraxella catarrhalis are the most common clinical manifestations in these patients, affecting 84–90% of CVID patients [Citation1,Citation3,Citation4]. In addition, a large proportion of CVID patients have clinical manifestation of non-resolving inflammation and autoimmunity, e.g. lymphadenopathy, splenomegaly, granulomas, cytopenia and interstitial lung disease. These, mostly sterile manifestations, involve not only B cell, but also immune cells such as T cells, monocyte/macrophages and dendritic cells [Citation5,Citation6]. Importantly, CVID patients with these noninfectious complications seem to have a higher mortality than those with ‘infection only’ [Citation7,Citation8].
One of the most commonly affected organs in CVID is the gastrointestinal (GI) tract [Citation9–11], involving infectious and noninfectious complications. The GI tract have the largest immune organ in the body [Citation12], and therefore it can be anticipated that an immunodeficiency such as CVID may influence the microenvironment in the GI tract. Although CVID patients are more prone to GI infections (e.g. Giardia (G) lamblia, certain Salmonella species and Campylobacter jejuni) [Citation3,Citation13], it is not the immunodeficiency per se that causes the most GI disease in CVID, but the wide spectrum of noninfectious GI pathology [Citation11]. The etiology of CVID enteropathy is multifactorial (), and in this review, we will focus on the noninfectious GI inflammation in CVID, potential consequences, and future perspectives.
2. CVID enteropathy – a wide spectrum of symptoms and severit
2.1. What is ‘CVID enteropathy’
The prevalence of GI disease/enteropathy is estimated to be 9–34% in CVID patients [Citation7,Citation9,Citation11,Citation14,Citation15]. These studies include cohorts with different diagnostic criteria for CVID enteropathy/GI disease. The lack of a universal definition illustrates one of the main challenges of CVID enteropathy. Whether or not to use histopathological- or symptomatic-based definitions, or a combination of both, has not reached a consensus. Exclusion of GI infections is included in some definitions, but not all. shows some of the definitions used/suggested for CVID enteropathy. This lack of consensus is problematic not only for the description of the prevalence of enteropathy, but also when comparing studies and conducting research. In mechanistic studies, it is essential to define the disease phenotype as accurately and specific as possible. A wide disease definition will increase both the genetic and environmental heterogeneity within the group and the likelihood of detecting the underlying causal factors, may drastically decrease. In the end, the lack of homogeneity, within the group studied, will have implications for finding targeted therapy for these patients.
Figure 1. CVID enteropathy. A suggested etiology model of CVID enteropathy where the size of the circles reflects the suggested impact on the complex enteropathy phenotype.
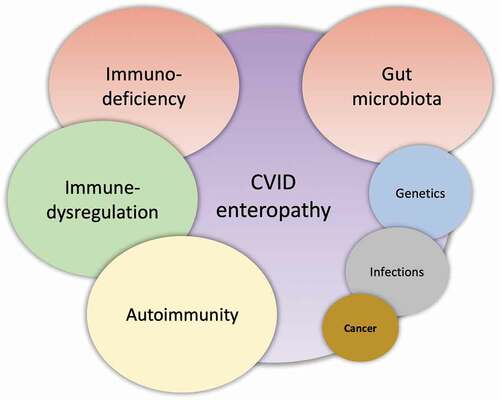
Table 1. An overview of some of the definitions used for CVID enteropathy
Intermittent or persistent diarrhea has been described as the most common GI symptom and disease manifestations in CVID patients, reported in 9–60% of cases [Citation4,Citation11,Citation13,Citation14,Citation16–18]. The large variation of prevalence in these reports may be ascribed to unbiased study population, retrospective rather than cross-sectional study design and a low number of participants in some of the studies. Prospective studies are scarce. In a cross-sectional study including 103 unselected CVID patients, we found that the most common GI symptoms were bloating (34%), followed by pain (30%) and diarrhea (26%) [Citation19]. It is important to distinguish reported ‘symptomatic’ GI disease from GI inflammation as some CVID patients may have GI symptoms without GI inflammation and asymptomatic CVID patients may have GI inflammation [Citation19,Citation20].
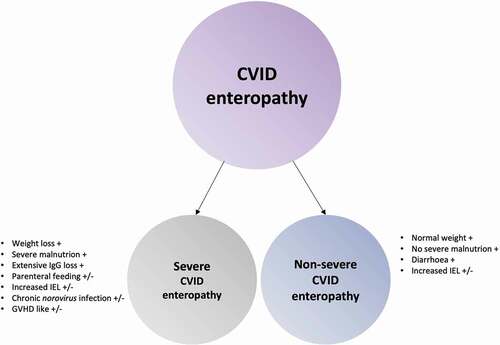
Sifers et al. described a subset of CVID patients that developed a more severe CVID-associated chronic enteropathy. These patients were characterized by weight loss, protein malnutrition, and in some, prolonged parenteral feeding [Citation21]. This sub-group of patients should be identified and followed closely by the clinicians and dieticians, to avoid secondary complications of malnutrition. These patients often have increased intra-epithelial lymphocytes (IEL), positive norovirus PCR in stools/mucosal samples and an extensive IgG-loss through the gut [Citation21–23]. In addition, some of these patients show histopathological changes in the GI tract similar to Graft versus host disease (GVHD) [Citation24], normally found as a complication to organ/BM transplantation. We suggest that these patients are referred to as Severe CVID enteropathy, whereas the patients with intermittent/chronic diarrhea without weight loss, malnutrition and severe GI-loss are referred to as Non-severe CVID enteropathy ().
Figure 2. CVID enteropathy sub-classes. CVID enteropathy divided into the two subclasses: Severe CVID enteropathy and Non-severe CVID enteropathy.
2.2. Norovirus infection as a cause of CVID enteropathy?
In 2015, Woodward et al. published a study of eight CVID patients with severe enteropathy, weight loss, and malnutrition that had positive norovirus PCR. They suggested that chronic norovirus infection could be the cause of CVID enteropathy [Citation23]. As a direct result of this publication, we tested for norovirus RNA in mucosal biopsies from 52 CVID patients (135 biopsies) from three different sites – stomach, duodenum and colon by PCR [Citation19]. We did, however, not find any positive biopsies for norovirus, although 47% suffered from one or more predefined GI symptom, of which 26% had diarrhea. We concluded that norovirus is not a common cause of CVID enteropathy. However, the cohort described by Woodward et al. appears to be the same subgroup described above as CVID-associated Severe CVID enteropathy characterized by weight loss and malnutrition. Similarly, in a small study exploring chronic norovirus infection in patients with primary immunodeficiency, norovirus infection was associated with low BMI, high IgG requirements, villous atrophy, increased IEL, absence of plasma cells and GVHD-like findings in the gut [Citation25]. The question is if the presence of norovirus in the GI tract of these patients is a ‘red herring’, and not the main cause of the inflammation. Instead, the presence of norovirus is merely a (bio)marker of severe immunodeficiency and in particular GI inflammation in CVID. In one recent case report, an undiagnosed CVID patient suffering from enteropathy-related malabsorption and chronic norovirus infection, was treated by IVIG alone, with complete resolution of GI symptoms and weight gain, suggesting that correction of the immunodeficiency may be sufficient in some cases [Citation26]. However, for most CVID patients, IVIG alone rarely cures the norovirus infection [Citation27,Citation28]. Antiviral therapy with ribavirin has been tried to eradicate the norovirus infection and thereby improve GI symptoms and inflammation. Woodward et al. described successful outcomes of ribavirin therapy in two patients and no effect in three patients [Citation23]. Following this observation there have been published several case-reports of CVID patients that did not have effect of ribavirin [Citation21,Citation27,Citation28]. Also, nitazoxanide, primarily used for the treatment of G. lamblia and cryptosporidiosis infections, has been proposed as anti-viral therapy for norovirus infection in CVID, but there is no data supporting this proposal [Citation27]. Additionally, there are case reports of pleconaril (an antiviral medication that has been used for the treatment of enterovirus infection [Citation29]), supportive therapy with human breast milk and enteral/oral immunoglobulin [Citation27,Citation28], all of which did not have any effect on the severity of symptoms. However, prednisone 1.5 mg/kg significantly improved the GI symptoms in one patient [Citation28]. Furthermore, patients with chronic norovirus infection and increased IEL/villous atrophy did not respond to a gluten-free diet (GFD) [Citation14,Citation23], suggesting another cause of increased IEL than gluten in these patients. Of note, all these studies are case-reports, and no systematic studies have been done regarding norovirus and CVID patients. The results should therefore be interpreted with caution.
We suggest that chronic norovirus infection in the gut is as a consequence of severe chronic GI inflammation in CVID, and not the main cause of the inflammation and GI symptoms.
3. CVID and upper GI
3.1. Chronic gastritis
Atrophic gastritis is reported in 3–20% of CVID patients [Citation8,Citation14,Citation19,Citation30] (). In our cross-sectional study including 50 Norwegian CVID patients, who were both symptomatic and asymptomatic, 45% of patients had inflammation of the stomach, of which 18% had chronic gastritis, not associated with Helicobacter (H.) pylori, EBV, or CMV infection [Citation19]. In a cohort of 30 French CVID patients with GI symptoms, macroscopic abnormalities in the stomach were found in 77% of patients, which included aspects of erythematous (27%), follicular (13%), atrophic (27%), and ulcerative (10%) gastritis [Citation31]. In a Finnish study, including 71 patients, chronic active gastritis was found in 15% and atrophic gastritis in 17% [Citation14]. In both studies, acute/chronic gastritis was infrequently linked to H. pylori infection, but H. pylori infection was uncommon in the Finnish cohort (6%), compared to the French study (27%) [Citation14,Citation31]. Furthermore, in an Australian cohort, only 9% of CVID patients were H. pylori positive, although 34% of the patients had gastric histology such as intestinal metaplasia (20%), atrophic gastritis (8%) and moderate to severe non-atrophic gastritis (16%) [Citation32], and in a Dutch study H. pylori was found in 10% and gastritis in 20% [Citation33]. Point being, that although H. pylori infection has previously been associated with gastritis in CVID [Citation34], studies from countries where H. pylori is less prevalent implies that H. pylori is not the only contributor to the risk of developing gastritis. Interestingly, Malamut et al. showed that in three patients with severe gastric corpus atrophy and H. pylori, persistent gastric atrophy with intestinal metaplasia consisted three years after eradication of H. pylori (H. pylori was absent on control endoscopy) [Citation31], suggesting that continuous mucosal processes occur unrelated to the presence of H. pylori. Thus, although it is likely that H. pylori can act as a trigger or catalyst of the inflammatory process in effected individuals, immune dysregulating processes unrelated to infection is probably equally important.
Increased incidence of stomach cancer in CVID has been associated with increased incidence of atrophic gastritis and intestinal metaplasia [Citation4,Citation13,Citation32,Citation35] which will be discussed later.
3.2. Celiac-like disease in CVID
3.2.1. The controversy of celiac disease in CVID
The most consistent histological finding in the upper GI tract in patients with CVID is very similar to that of celiac disease () [Citation19,Citation24,Citation31]. The histological findings include lamina propria mononuclear cell infiltration, and increased number of IEL with or without, villous atrophy. However, compared to celiac disease, biopsies from CVID patients often show lower IEL, and villous atrophy, if present, may be more partial than severe. Furthermore, acute duodenitis characterized by strong neutrophil infiltration and follicular lymphoid hyperplasia may be present, which are unusual in celiac disease [Citation31]. It has been suggested that celiac disease differs from increased IEL in CVID with regards to some natural killer cell markers, and the TCF-gamma/delta phenotype characteristic that is generally not present in CVID [Citation31]. Also, Venhoff et al suggested that CVID patients with celiac-like disease had expansion of CD21low B-cells [Citation36], but we did not find any association with CD21low B-cells and increased IEL in 50 CVID patients [Citation19]. The expansion of CD21 low B-cells reported by Venhoff et al. is probably due to the overlap with autoimmune disease and not celiac disease specific. The major histological feature separating celiac disease from CVID is the lack of plasma cell infiltration [Citation24,Citation37]. The question is if this represents ‘true’ celiac disease triggered by gluten or not.
Figure 3. Macroscopic and microscopic gastrointestinal pathology and gut microbial dysbiosis in CVID. Gastrointestinal disease in CVID is associated with immune dysregulation, immunodeficiency and autoimmunity that effects the stomach, liver and large- and small intestine. The gut microbial composition in CVID is unhealthy and together with GI inflammation it allows gut leakage of microbial products into the blood stream to initiate/maintain an inflammatory process.
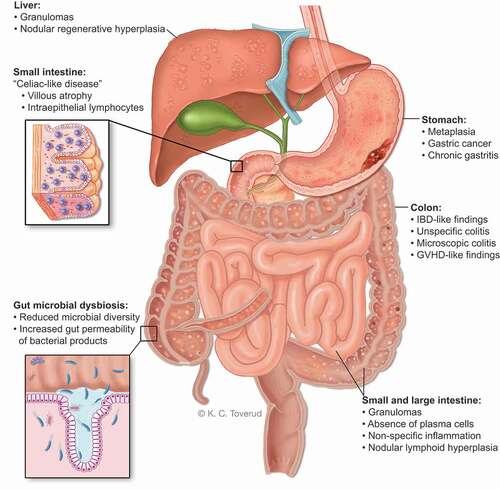
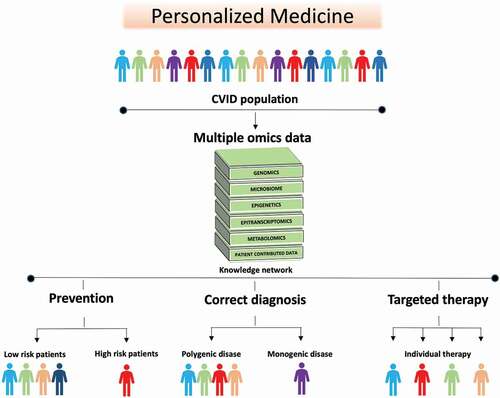
Figure 4. The future of CVID is personalized medicine. The heterogenous nature of CVID and the overlap between CVID enteropathy and inflammation/autoimmunity in other organs makes it logical to approach personalized therapy in CVID as a group. Data such as genomics, transcriptomics, metabolomics, and metagenomics including samples from blood, tissues and feces, are combined. Then statistical and mathematical integration methods for multiple omics datasets are applied to unravel the complexity of biological systems involved in CVID and to identify signatures that can stratify the patients further. This new knowledge can be used in: (i) Prevention, to detect patients at high risk of developing complications e.g. gastric cancer, liver failure or severe chronic enteropathy (ii) Correct diagnosis: to identify monogenic disease that enables individualized treatment strategy, and individuals that are wrongly categorized as CVID (iii) Targeted therapy: to predict which patients are more likely to respond to specific therapies e.g. biological therapy directed at specific mechanistic pathways, nutritional intervention e.g. high fiber diet, microbiome treatment e.g. probiotics, or a combination thereof.
In a cross-sectional study, including both symptomatic and asymptomatic CVID patients, we found increased IEL in the duodenum (pars descendens) in 46% of CVID patients. Histopathological findings of increased IEL did not correlate with GI symptoms. Only one sample had villous atrophy, 2%, but this is in line with the one other published cross-sectional study of GI pathology in CVID [Citation30]. However, retrospective studies have shown a higher prevalence of villous atrophy in CVID 30–50% [Citation31,Citation38], including patients with only GI symptoms and/or anemia. The higher prevalence in the retrospective studies may be due to study design and selection bias, i.e. biopsies originating from patients with more severe symptoms sampled over several years. However, in a recent large retrospective study from Finland, Pikkarainen et al. found villous blunting and increased IEL in only 10% (7/71) of CVID patients with GI symptoms [Citation14]. An accurate estimate of villous atrophy in CVID is limited by small studies and the fact that most studies are retrospective.
Venhoff et al. also explored the association between the celiac disease related HLA profiles and celiac disease-like findings in duodenal biopsies in 20 patients. All patients had increased IEL and diarrhea (chronic or recurrent), and 75% had villous atrophy, whereas half of the patients had malabsorption. Only four of these (20%) carried the celiac disease related HLA profile [Citation36], suggesting limited association between celiac disease related HLA profile and histopathological changes resembling celiac disease in symptomatic CVID patients. Likewise, in a larger cohort (n = 50), we found no correlation between the histological findings resembling celiac disease and the celiac disease related HLA profiles, HLA-DQ2 and HLA-DQ8. Furthermore, when comparing gene-expression analysis from duodenal biopsies, we found that CVID patients with ‘celiac-like disease’ and true celiac disease were different [Citation19], suggesting that these similar histological findings represent different disease mechanisms and disease entities.
Earlier, it has been postulated that G. lamblia infection in CVID was associated with increased IEL in duodenum [Citation22]. A concurrent infection with G. lamblia and increased IEL was found in 23% (7/31) [Citation31], 5% (1/20) [Citation36] and 0% (0/23) [Citation19], in three different studies. The discrepancy reported in these studies is probably related to the prevalence of G. lamblia in the country studied, and for the majority of CVID patients increased IEL or lymphoid hyperplasia are not a related to G. lamblia.
3.2.2. GFD, not indicated in CVID
There is an inconsistency in the reported literature both in terms of histological and symptomatic response to GFD [Citation14,Citation24,Citation31,Citation36]. In a study by Malamut et al. only two of 12 CVID patients with villous atrophy responded to a GFD [Citation31]. Some advocate that HLA genotyping for HLA 2.5 and HLADQ8 can be useful in selecting the CVID patients with celiac-like histopathology who should try a GFD. In the study mentioned above by Venhoff et al., two out of four CVID patients with increased IEL, villous atrophy and HLA 2.5 or HLADQ8 had a histological response to a GFD [Citation36]. Also, Pikkarainen et al. demonstrated an inconsistency in HLA associated celiac disease profile, increased IEL with villous atrophy and response to GFD [Citation14]. The improvement of duodenal biopsies 3–6 months after initiating GFD, and ideally the return of the histopathological findings after re-introduction are the only certain/definitive evaluations of response to GFD in CVID. The symptomatic relief experienced by some CVID patients after introducing GFD [Citation39], may be due to alteration of microbial composition secondary to alteration of diet, natural fluctuations in symptoms (e.g. intermittent diarrhea) or a placebo effect. Also, in the general population, individuals without celiac disease may experience relief of GI symptoms after self-initiation of GFD. In fact, most Americans on GFD does not have celiac disease, illustrating a large cohort of individuals that subjectively experience symptomatic relief on a GFD [Citation40]. We do not recommend GFD to our patients because our interpretations of the current literature are that increased IEL in CVID patients represents a general immune dysregulation, also affecting the small intestine, and not gluten sensitivity [Citation19,Citation41]. This is also supported by case-reports where CVID patients with celiac disease-like histopathology experienced mucosal response after starting corticosteroid therapy [Citation24]. The initiation of GFD in CVID patients is not without potential complications as dietary restrictions can cause macro- and micronutrient deficiencies [Citation42], which may be particularly challenging in the patients with preexisting malnutrition secondary to chronic diarrhea [Citation43]. In our opinion, one should be particularly careful with patients having Severe CVID enteropathy with known malnutrition and weight loss (see above).
4. CVID and lower-GI
4.1. IBD-like disease, microscopic-, and unspecific colitis
The GI inflammation observed in CVID is very heterogenous. Colon biopsies from CVID patients have a wide range of morphological findings (). Histological findings may be ‘IBD-like’ with intraepithelial or sub-epithelial lymphocytosis, prominent apoptosis, granulomas, and crypt distortion, accompanied by the absence of plasma cells in the lamina propria [Citation44] or it can mimic histological features seen in lymphocytic/collagenous colitis, and/or gut involvement resembling GVHD (as discussed above) [Citation18,Citation22,Citation45].
The prevalence of inflammatory bowel disease (IBD) in CVID varies and has been reported in 2–14% [Citation13,Citation14,Citation19,Citation31,Citation46–48], dependent on the cohort studied and if both symptomatic and asymptomatic patients are included. Microscopic colitis, lymphocytic or collagenous, are found in 10–26% patients of CVID [Citation14,Citation19,Citation31]. These patients may macroscopically have normal colon, but biopsies may reveal microscopic colitis. Therefore, in CVID patients with GI symptoms, biopsies should always be taken, even though it macroscopically appears normal. Compared to the diagnostic criteria for the various GI-disease entities, CVID often lack some diagnostic criteria, and an unspecific colitis is annotated.
In a recent Finnish study, unspecific colitis was found in 14%, whereas Malamut et al. described 23% with unspecific colitis, among CVID patients with GI symptoms [Citation14,Citation31]. The unspecific colitis was more common than IBD in these studies [Citation14,Citation31], supporting that the colitis in CVID is often nonspecific. Interestingly, in the Finnish study, all patients that had colonic inflammation also had duodenal inflammation, suggesting a more general state of GI inflammation as described above [Citation14].
GI inflammation in CVID has been associated with different patterns of inflammation compared to IBD [Citation49]. For example, one study found that the mononuclear cells in the lamina propria of CVID patients produced significantly more IL-12 and IFN-y, but not IL-23 and IL-1, compared to classical Crohn’s colitis, suggesting an alternative pathway of inflammation in CVID compared to IBD [Citation20,Citation50]. Thus, in our opinion, true IBD is rare in CVID, and in addition to lymphocytic colitis that is seen in 8–17% [Citation14,Citation19,Citation31], biopsies most often reveal unspecific inflammation.
Granulomatous disease occurs in 8–22% of CVID patients and may affect many organs including the GI tract and liver [Citation51,Citation52]. In some cases, it has been associated with diarrhea and weight-loss, and therapy can be difficult [Citation51].
4.2. Liver inflammation in CVID
The reported prevalence of liver involvement in CVID varies from 3% to 79% [Citation53,Citation54], reflecting different study design and inclusion criteria. Nodular regenerative hyperplasia (NRH) is the most common form of liver involvement in CVID [Citation2], and may represent an immune-mediated manifestation in itself [Citation55]. The pathogenesis of NRH is not fully understood, but infiltration of CD8+ cytotoxic T-cells in the liver sinusoids, with evidence of apoptotic injury to the sinusoidal endothelial cells, has been described and may indicate an autoimmune and possibly T-cell driven origin for NRH [Citation56,Citation57]. Likewise, increased CD8 + T-cells in the liver parenchyma with increased production of liver interferon-ɣ and CD8 + T- has also been reported, leading to cell mediated hepatocyte loss, regenerative process and ultimately vascular abnormalities [Citation58]. The nodular areas are thought to be a hypertrophic response to either normal or increased perfusion [Citation59], and may sequentially exert compression of the hepatic sinusoids, which in turn leads to perisinusoidal fibrosis [Citation55]. Only a few studies have reported prevalence of bioptically verified NRH among CVID patients, with prevalence varying from 41% to 87% [Citation54,Citation57,Citation60,Citation61]. In contrast, NRH is a relatively rare finding in the general population, affecting only 0.5–2.6% [Citation59], and the majority of cases are associated with systemic disorders, e.g. rheumatoid arthritis and systemic lupus erythematosus [Citation62–64]. NRH has the capacity to progress, resulting in portal hypertension, splenomegaly, ascites, varices, and liver synthetic dysfunction in CVID [Citation58], but may also present with dyspnea, accompanied by hypoxia, secondary to hepatopulmonary shunting [Citation61]. Notably, NRH can be associated with granuloma formation underscoring that the liver NRH could be immune-driven and further research should examine if immune-modulatory therapy could attenuate the progression of this liver disorder.
Cases of combined CVID with autoimmune hepatitis [Citation44,Citation65,Citation66], primary biliary cholangitis [Citation13] and primary sclerosing cholangitis [Citation67] have also been described.
As of now, liver transplantation remains the only treatment option in end-stage liver disease. Limited evidence, in a CVID setting, has been published, with a total of 35 cases worldwide [Citation11,Citation61,Citation68–76], with increased survival rates in the last decade [Citation61]. Data regarding recurrent disease after liver transplantation is limited, but relapses of autoimmune liver disease [Citation71] and NRH have been documented [Citation69].
5. Common features of upper and lower GI
5.1. Immune dysregulation is related to reduced plasma cells in GI biopsies
Co-existence of CVID with GI-symptoms and autoimmune manifestations in other organs suggests that the GI inflammation is a manifestation of a more general phenomenon characterized by immune dysregulation [Citation18,Citation31]. A reduced number of plasma cells in mucosal tissue is the most common histopathological finding in GI biopsies from CVID patients varying from 42% to 83% in duodenum [Citation19,Citation31,Citation37,Citation77] to 56–63% in the colon [Citation31,Citation37]. Reduced number of plasma cells in GI biopsies is associated with increased plasma levels of monocytes and T-cell activation and with certain B-cell phenotypes in peripheral blood, i.e., decreased class-switched memory and increased CD21low B cells [Citation19]. The finding of reduced plasma cells in mucosal tissue could therefore potentially be used as an indicator of a more ‘inflammatory phenotype,’ a state of low-grade systemic inflammation and immune activation, which in turn is related to inflammatory and autoimmune complications [Citation19].
5.2. Nodular lymphoid hyperplasia
Nodular lymphoid hyperplasia is a common finding in the gut of CVID patients () detected in 38–53% of CVID patients in two cross-sectional studies, including both symptomatic and un-symptomatic patients [Citation19,Citation30]. Nodular lymphoid hyperplasia was associated with increased numbers of circulating B-cells, but not GI symptoms [Citation19]. Both the cause and consequences of lymphoid hyperplasia are unknown. An association between lymphoid hyperplasia and G. lamblia has been suggested [Citation22], but lymphoid hyperplasia was frequently found (38%) in a cohort where G. lamblia was tested, but not detected [Citation19]. We suggest that lymphoid hyperplasia in the gut is not separate from lymphoid hyperplasia in lymph nodes or in other organs, e.g. the spleen, and is a clinical manifestation of immune dysregulation and not related to GI infections.
6. Immunosuppressive therapy
There are very few, if any, randomized controlled trials evaluating the effectiveness of different treatment options for CVID enteropathy. This is mainly due to the difficulties in gathering large enough cohorts, and the heterogeneity within the CVID enteropathy group. Therefore, for CVID patients with IBD-like disease, most centers advocate the same therapy as for immunocompetent patients [Citation1]. This includes steroids, 5-aminosalisalicylic acid, 6-mercaptopurine and azathioprine [Citation31,Citation44], but individuals with CVID appear to be more refractory to treatment [Citation78], perhaps reflecting different pathways of inflammation compared to IBD [Citation20]. CVID patients with noninfectious non-IBD enteropathy can respond to steroids [Citation31], but high dose treatment (i.e. Prednisolone ≥ 30 mg a day) may be problematic due to an increased risk of opportunistic infections [Citation44]. Budesonide treatment produces less systemic steroid effects, but the reduction of dosage from 9 milligram (mg) to 6 mg or 3 mg (which is a recommended dose reduction) often, in our experience, leads to a relapse of symptoms. A clinically favorable outcome after treatment with anti-tumor necrosis factor alpha (anti-TNF-α) inhibitor, either alone or in combination with azathioprine, has been reported in a small number of cases [Citation79–81], however histological improvement after anti-TNF-α treatment has not been demonstrated. Other studies have reported no clinical improvement after anti-TNF α therapy or Cyclosporine orally [Citation31].Three studies with a total of eleven patients receiving vedolizumab demonstrated effect in 4/11 (36%) [Citation21,Citation82,Citation83], however the studies included patients with different histopathological findings and severity of disease. Overall, for many patients with CVID enteropathy, medical therapy is often unsatisfactory or transient.
The detection of a monogenic cause of CVID enteropathy may aid the selection of therapy. For an example, a patient with a CTLA4 mutation had improvement with vedolizumab [Citation84], whereas two patients with BACH2 and NFKB1 mutations, respectively, had adverse reactions to the same therapy [Citation21]. The underlying genetic factors in these subjects may have led to the difference in response to vedolizumab therapy. For patients with CTLA4 deficiency and enteropathy, tailored therapy with abatacept, should probably be the first-line steroid-sparing agent [Citation85]. A correct molecular diagnosis is essential not only with regards to targeted therapy, but also for consideration/timing of HSCT if the immune dysregulation is severe/resistant to therapy.
7. GI inflammation and link to GI cancer
CVID patients have an increased incidence of gastric carcinoma [Citation86]. In the largest study so far, consisting of 455 Italian CVID patients, Pulvirenti et al. found that the risk for gastric cancer was increased 6.4-fold based on 25 cases [Citation87].
A progressive disease model known as Correa’s cascade, from active gastritis via gastric atrophy, intestinal metaplasia, dysplasia to carcinoma, has been established in the general population for many years [Citation88], with H. pylori infection being the most important risk factor [Citation89]. Quinti et al. reported intestinal metaplasia to be a common finding in an Italian cohort of CVID patients over the age of 18 undergoing biannual gastroscopy [Citation4]. They also found that progression to atrophic gastritis and malignancy seemed to develop at a higher rate in H. pylori positive CVID patients compared to the general population. Pulvirenti et al. found only an invariable association with chronic atrophic gastritis and extensive intestinal metaplasia, and only 3/7 had positive H. pylori. Interestingly, in two patients the cancer developed rapidly without signs of dysplasia on gastroscopies one year before a high grade gastric cancer [Citation87], indicating that gastric cancer development may not follow the traditional Correa’s cascade. Furthermore, CVID patients were younger compared to the reference population at the time of their gastric cancer diagnosis, and these adenocarcinomas were of intestinal type [Citation87,Citation90]. The biopsies showed increased numbers of intra-tumoral lymphocytes, accompanied by typical CVID features such as reduced number of plasma cells and nodular lymphoid hyperplasia, features suggestive of chronic inflammation of the gastric mucosa [Citation87,Citation90].
Gullo et al. compared the immune infiltration in adjacent/distant mucosa to the tumoral gastric tissue from CVID and non-CVID patients. By using immunohistochemistry and subsequent automatic quantification analyses they found higher rates of CD4+, CD8+, Foxp3+, GATA3+ and PD-L1+ immune cells and, as expected, reduced CD20 + B-lymphocytes and CD138+ plasma cells in CVID patients compared to non-CVID patients [Citation91]. However, a major confounding factor in this study is that the majority of CVID patients with gastric cancer (n = 8/9) had H. Pylori infection and the mucosal immune activation could therefore be secondary to infection.
Most of the published CVID cohorts investigating GI cancer in CVID, are not tested for monogenic cause of CVID. This may be important as CVID cohorts may contain individuals with monogenic disease that are predisposed to gastric cancer. For example, NF- kB1 deficiency is the most common monogenic cause of CVID [Citation92], and recently Reilly et al. showed that a mouse model of NF- kB1 deficiency causes gastric cancer by dysregulating inflammation and immune check points through a STAT1-dependent process [Citation93]. Also, CVID patients with STAT1- gain of function variants may present with a phenotype resembling CVID [Citation92]. The point is that recognizing monogenic cause within a CVID cohort, may also identify patients at risk for developing gastric cancer, allowing better surveillance of high-risk patients.
That aside, there is probably an increased risk of gastric cancer outside monogenic disease in CVID. We believe that immune dysregulation may play an important role in the development of gastric cancer in CVID. It is evident that the increased burden of DNA damage typically occurs in conditions characterized by persistent inflammation and oxidative stress, which are both characteristic features of CVID [Citation94]. Epigenetic modulation, which may affect immune checkpoints and the signaling network that controls inflammation, may ‘transfer’ chronic inflammation into cancer in CVID.
Extranodal marginal zone lymphoma in the gut mucosa is rare but has been described in case reports in CVID [Citation95–98].
8. The contribution of gut microbiota and mucosal IgA to gut inflammation
8.1. Reduced microbial diversity and a CVID-specific dysbiosis index
In the last decade, the interaction between gut microbiota and intestinal and systemic inflammation has received much attention as a possible pathogenic mechanism in several autoimmune and immune-mediated disorders [Citation99–101]. However, there are only a few studies that have explored the role of gut microbiota in CVID [Citation102–111], with a limited number of patients in many of the studies. The first study was published in 2016, where we showed that the gut microbiota of CVID patients differed from healthy individuals, with both reduced microbial diversity and altered microbial composition. We also calculated a CVID-specific dysbiosis index that captured the dysbiosis seen in CVID [Citation102,Citation109]. This index consisted of ten taxa: Bacilli, Dorea, Roseburia, Gammaproteobacteria (increased in CVID), and Bifidobacterium, Odoribacteracea, Christensenellaceae, Blautia, Sutterella, Desulfovibrionacea (reduced in CVID). Furthermore, the CVID dysbiosis index showed a strong correlation with systemic inflammatory markers indicating T-cell activation and monocyte/macrophage activation [Citation102]. Later, in a an expanded cohort of CVID patients using a different statistical method exploring taxa on genus level, we found five taxa that differentiated CVID and heathy controls: Christensenellaceae R-7 group (reduced in CVID) and Hungatella, Flavonifractor, Veillonella and Escherichia-Shigella (increased in CVID) [Citation108]. Recently, the increased presence of Hungatella in CVID patients, was confirmed in a metagenomic study of six CVID patients compared to household controls [Citation111]. With regards to clinical subgroups, CVID patients with inflammatory and autoimmune complications (‘Complications’) have significantly reduced microbial diversity compared to CVID patients with only infections (‘Infection only’) [Citation102,Citation106]. However, there was no difference in microbial diversity between CVID patients with enteropathy compared to those without [Citation104,Citation108]. This may be due to a considerable overlap between the different clinical subgroups in CVID, e.g. CVID patients without enteropathy often have other inflammatory and autoimmune complications, which may affect the gut microbial composition. This clinical overlap may contribute to the lack of statistical significance when comparing clinical subgroups, except for Infection only and Complications which are distinct subgroups and have no overlap.
The majority of gut microbiota studies in CVID are performed on stool samples, but the small intestinal microenvironment is probably also important for absorption of nutrients/micronutrients and gut microbial leakage mechanisms. In a recent study, using hydrogen breath test, small intestinal bacterial overgrowth was found in approximately 40% of CVID patients and was associated with GI symptoms such as bloating [Citation112]. However, Shulzhenko et al. have studied the duodenal microbiome in more detail using next-generation sequencing, and they did not observe significant differences in bacterial abundance in duodenal biopsies from CVID patients compared to control groups [Citation104]. More studies are needed to characterize the microbial community in the small intestine and how it may contribute to the CVID phenotype.
8.2. Microbial products from the gut can initiate systemic inflammatory responses
It is evident that microbial products originating from the microbial composition in the gut can occur in the blood stream and initiate inflammatory processes. One possible mechanism is directly linked to gut inflammation: When tight junctions between epithelial cells in the intestinal mucosa are compromised, e.g. due to gut inflammation, influx of bacterial products and other foreign substances can occur. As an example lipopolysaccharides (LPS), a component of the gram negative gut wall, may, through gut leakage mechanisms, leave the GI tract and subsequently interact with TLRs, and related molecules in the blood stream, and consequently lead to activation of innate immunity [Citation113]. While the gut microbiota can initiate systemic inflammation, it may also activate the inflammatory pathways locally in the GI tract, including activation of inflammasomes in the intestinal wall, e.g., nucleotide-binding domain and leucine-rich-repeat-containing proteins NLRP3 and NLRP6, through the effector proteins IL-18 and IL-1b [Citation114]. The altered gut microbiota may also stimulate certain inflammatory cytokines such as CCL5, which can result in increased permeability and influx of microbial components. These bidirectional interactions may represent an inflammatory pathogenic loop with gut microbiota as a central regulator of GI and systemic inflammation as an outcome. Both gram negative bacteria and LPS are increased in CVID and correlate with each other and inflammatory markers [Citation102,Citation115]. The gut microbiota dependent metabolite trimethylamine N-oxide (TMAO) has been linked to systemic inflammation in other diseases, e.g. HIV and cardiovascular disease [Citation116,Citation117]. The role of TMAO in CVID and its relation to diet, gut microbiota and inflammation was recently reported. We found that TMAO was increased in CVID compared to controls, and that this increase was associated with increased plasma levels of inflammatory markers (TNF and IL12) and LPS, as well as increased Gammaproteobacteria in stools [Citation115]. The association of TMAO with Gammaproteobacteria is interesting as this is an important taxon responsible for gut microbial dysbiosis in CVID [Citation102,Citation108]. Whether or not TMAO is merely a biomarker of inflammation or also a mediator of inflammation contributing to systemic inflammation in CVID through the gut-microbial axis is not clear [Citation118]. However, mechanistic studies have shown that TMAO triggers activation of the NLRP3 inflammasome in endothelial cells, involving increased production of reactive oxygen species, suggesting a role of TMAO in inflammation beyond just a biomarker of inflammation [Citation119,Citation120]. Of note, we found that the consumption of red wine had a potential beneficial effect on reducing TMAO. The negative correlation between consumption of red wine and plasma TMAO may be related to the fact that red wine contains compounds that inhibit trimethylamine formation, which is a TMAO precursor. Further studies are needed to explore this association.
8.3. Targeting CVID gut microbiota
A recent study of B-cell deficient mice (CD19−/−) showed reduced anti-commensal IgA responses and an overgrowth of anaerobic bacteria that corresponded with the development of an inflammatory enteropathy in the ileum. Interestingly, the enteropathy found in these mice was reversed by both an antibiotic (metronidazole targeting anaerobic bacteria) and a GFD (in separate experiments), illustrating an important proof of concept in that altering gut microbial composition, via diet or antibiotics, may affect GI inflammation [Citation121]. It is important to emphasize that this is not a CVID mouse model and the results from this study cannot be transferred to human CVID patients, as previously discussed [Citation122].
As of now, there is only one study in CVID targeting gut microbiota composition. In this study, we used rifaximin a broad-spectrum antibiotic acting locally in the gut, with negligible systemic absorption. Rifaximin is approved for travelers’ diarrhea and is used for prevention of liver encephalopathy (reduce LPS). Rifaximin has been used in CVID enteropathy for small intestine bacterial overgrowth [Citation123], but randomized studies have not been performed. We designed a proof-of-concept study to explore if alteration of the gut microbiota by rifaximin could reduce LSP (increased in CVID) and affect systemic inflammation. We found that, although the microbiota composition was significantly changed by rifaximin, there was no significant change in systemic inflammatory markers or LPS in CVID patients taking rifaximin. The reason for this may be that the important bacteria responsible for the CVID dysbiosis, i.e. the ten taxa in the CVID dysbiosis index, were not changed by rifaximin.
In terms of IVIG therapy, Fadlallah et al. showed that IVIG poorly targets CVID gut microbiota most likely because it does not contain dysbiosis-specific antibodies. This may explain why IVIG does not have an effect on GI symptoms in CVID [Citation105].
8.4. The role of mucosal IgA in CVID enteropathy
The gut microbiota is important for the secretion of IgA and the maturation of the immune system in the gut. This has been elegantly illustrated in mice studies where germ-free mice are deficient in secretory IgA until colonized with commensal bacteria. Only after the introduction of bacteria in the gut, the intestinal IgA response is induced [Citation124–127]. IgA is also important for mucosal defense against invading microbes, and for retaining a mutual beneficial microbial environment with the commensal bacteria, favoring microbes beneficial to a healthy gut environment [Citation128]. The association between reduced gut microbial diversity and serum IgA levels have been inconsistent in CVID [Citation102,Citation108]. A recent publication by Shulzhenko et al. shed some new light on this observation as they detected a discrepancy between serum IgA and the expression of mucosal IgA in CVID. The aim of their study was to explore if there was an association between IgA deficiency and enteropathy in CVID, but initially they found no difference in serum IgA levels in patients with or without enteropathy. However, when exploring mRNA levels of IgA subclasses (encoded by the IGHA1 and IGHA2 genes) in the gut mucosa from CVID patients, they found that CVID patients with enteropathy had reduced expression of IgA subclasses in the mucosa compared to CVID patients without enteropathy. Furthermore, reduced IgA expression in duodenal biopsies was linked to increased occurrences of certain bacteria (i.e., Acinetobacter baumannii) in the duodenal microbial community that induced an inflammatory phenotype in macrophages [Citation104]. These findings support a role of reduced intestinal, but not serum, IgA in CVID enteropathy and a possible link to systemic inflammation and gut microbial dysbiosis. Of note, mucosal IgA deficiency may also affect microbial composition at other sites than the gut. Increased bacterial load in oropharyngeal microbiota has been associated with serum IgA < 1 g/L and inflammatory lung disease in CVID patients.
9. Genetics and GI inflammation
According to a recent publication in Nature, including 443 patients with CVID, monogenic defects were found in ~10% of CVID patients [Citation92], suggesting that monogenic disease resembling CVID, may be hidden in a CVID cohort. The most common genetic variants with a clinical picture resembling CVID and enteropathy are in the genes CTLA4, LRBA, or PIK3CD [Citation92]. A high suspicion of underlying monogenic cause should be undertaken, particularly if symptoms have been present since early childhood or if multiple autoimmune manifestations are present. Depending on the availability of WES/WGS analyses, it is advisable to genetically test patients with enteropathy for monogenic primary immunodeficiency.
That aside, the majority of CVID patients probably have a polygenic risk profile, as seen with many other immune mediated diseases [Citation94]. Outside HLA only one susceptibility gene, CLEC16A, has been identified in CVID [Citation129]. Interestingly, clinical sub-group analyses of CLEC16A showed that the most strongly associated SNP (rs17806056) in the CLEC16A gene was significantly associated with enteropathy and not only autoimmune subtypes in CVID [Citation129], suggesting that CLEC16A may play a mechanistic role in CVID enteropathy. In addition to CVID, CLEC16A has also been associated with other autoimmune diseases including primary biliary cirrhosis, Crohn’s disease and celiac disease [Citation130–133]. The overlap of CLEC16A associations in CVID and other GI diseases, raise opportunities for exploring mechanisms underlying the autoimmune co-morbidity and the chronic GI inflammation observed in CVID.
10. Conclusion
There is a gut inflammation in CVID that is part of the general immune dysregulation affecting patients with CVID, irrespective and unrelated to GI infections. The most common upper GI inflammation is chronic gastritis, which may in rare cases develop to gastric cancer, and duodenal inflammation resembling celiac disease. The inflammation affecting the lower GI tract is heterogenous and often characterized as an unspecific colitis. Recent studies have suggested a role of gut microbiota in the etiology of CVID, and the reduction of mucosal IgA in CVID enteropathy. The heterogenous group of patients makes it difficult to find common grounds for treatment for the GI inflammation in CVID.
11. Expert opinion
The heterogeneity and unknown mechanism behind CVID enteropathy, and thereby the lack of effective treatment, is one of the key challenges in the field of CVID. There is a considerable burden of disease with major negative impact on the quality of life, morbidity, and mortality [Citation8,Citation110]. Therefore, it is of utmost importance to unveil new molecular pathways that have a major impact on the understanding of CVID enteropathy and inflammation. This will also open for personalized and targeted therapy of CVID patients based on genetics, epigenetics, and the elucidation of molecular pathways that are activated.
We advocate a symptomatic-based definition of CVID enteropathy with diarrhea for at least 3 months and an exclusion of infectious bacterial and parasite infection, but not norovirus (as this may be persistent and indicate a more severe disease). However, we suggest dividing into two separate sub-groups based on disease severity. One distinct clinical sub-group with weight loss, severe malnutrition and, sometimes, chronic norovirus infection. These clinical findings are often accompanied by GI biopsies showing increased IEL and GVHD-like changes. We suggest the labeling this group Severe CVID enteropathy. The group may need a more aggressive approach in terms of immune suppressive therapy and genetic testing for monogenic disease and should be studied separately in mechanistic and prospective studies. The remaining CVID patients with CVID enteropathy are referred to as Non-severe CVID enteropathy.
Advancements in genetics with the introduction of next-generation sequencing with reduction in cost and time processing these samples, have made these analyses more accessible to clinicians and patients in many countries. Individuals with monogenic disease not classified as CVID, may be hidden in the CVID cohorts. These new gene defects with interrupted equilibrium of the immune system, may provide new clues to the biological effects of the immune receptors or proteins within the human immune system. This may lead to new treatment options, particularly biological medications, tailored to the mechanisms affected by the mutation. The unknown genetic background for the majority of CVID patients may hide important mechanistic pathways that can aid therapy.
The striking difference in gene expression from biopsies taken from the duodenum from CVID patients with celiac-like disease and ‘true’ celiac disease suggests that although histopathological findings are similar, they represent different disease entities. The inconsistent link to the celiac disease associated HLA profile and response to GFD, further supports this notion. Histopathological celiac-like findings in CVID may, instead of being secondary to gluten, reflect an immune dysregulated phenotype in these patients with immunodeficiency coexisting with persistent immune activation and inflammation. Based on this hypothesis, the approach to treatment should be immunomodulatory rather than a GFD. Further research should explore in greater depth, the nature of the small intestine inflammation in CVID at a molecular level, where e.g. regulation of DNA (epigenetics) and regulation of mRNA (transcriptomics) may elucidate novel pathogenic mechanisms in CVID.
There is a considerable overlap between GI enteropathy and autoimmunity in CVID, and it is difficult to discuss GI inflammation separate from the rest of the inflammatory CVID phenotype when many patients have additional inflammation and/or autoimmunity in other organs. We propose that CVID immunopathogenesis involves an interplay of genes, environmental factors, and dysregulation of immune cells, where gut microbiota and GI inflammation can both be important contributors or endpoints to the systemic immune activation seen in CVID, as previously discussed [Citation94].
In the near future, implementation of omics technologies to unveil potentially new disease mechanisms in CVID should be undertaken. On a group level, there are huge possibilities in more complex omics analysis combining gut microbiota with other samples such as blood, biopsies, and diet, as has been done for other clinical phenotypes [Citation134]. These findings could generate follow-up studies in appropriate tissues and could ultimately lead to improved clinical practice.
The development of novel therapeutic option in other parts of clinical medicine such as cancer and autoimmune- and autoinflammatory disorders could be applied to the CVID population targeting relevant pathways that are elucidated through mechanistic studies in these patients. Such novel therapeutic approaches should be investigated in prospective and, in some cases, randomized control trials that are almost lacking in the research field of CVID. This requires, however, collaboration between different countries and research groups.
The fact that, individuals show great inter-individual variation in the gut microbiota composition [Citation135] also make way for opportunities of more personalized medicine. There are already studies that have used integration of microbiota profile and metadata in a machine learning model, to give tailored personalized advice [Citation136,Citation137]. Our predictions on the evolvement of the field, 10 years from now, include a scenario where clinical data, blood samples (containing specific biomarkers, and inflammatory markers), host genetics (including HLA profile), gut microbiota profile and RNA/DNA sequencing data from biopsies are fed into a mathematical algorithm, where the result of the analyses points to which therapy that is more likely to work for each individual patient (). This approach requires a multidisciplinary approach that not only includes dedicated researchers and clinicians, but individuals with hands-on knowledge of multi-level bioinformatics. Considering the heterogeneity of CVID enteropathy, personalized medicine is probably the future for these patients.
Article highlights
The discrepancy in the definition of CVID enteropathy is problematic in terms of mechanistic studies and therapy.
There is a distinct subgroup within CVID enteropathy characterized by malnutrition, weight loss, and often with positive norovirus PCR. We suggest the nomenclature Severe CVID enteropathy for these patients.
Duodenal biopsies resembling celiac disease are probably due to a more general inflammatory processes in CVID and not gluten, and a gluten-free diet is therefore not indicated.
Gut microbial dysbiosis and mucosal IgA are associated with CVID enteropathy and systemic inflammation in CVID.
Personalized medicine is probably the future for CVID enteropathy, with a more aggressive approach to therapy that also include biological medications.
Declaration of interest
S Jørgensen (project number 2019089) is funded by grants from the South-Eastern Norway Regional Health Authority. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
We thank K Toverud for professional support in developing of this review article. We also thank P Aukrust for reading through the manuscript and for giving valuable feedback.
Additional information
Funding
References
- Cunningham-Rundles C. How I treat common variable immune deficiency. Blood. 2010;116:7–15.
- Bonilla FA, Barlan I, Chapel H, et al. International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4:38–59.
- Oksenhendler E, Gérard L, Fieschi C, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46:1547–1554.
- Quinti I, Soresina A, and Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007; 27: 308–316.
- Vinh DC, Patel SY, Uzel G, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115(8):1519–1529.
- Sharifi L, Tavakolinia N, Kiaee F, et al. A review on defects of dendritic cells in common variable immunodeficiency. Endocr Metab Immune Disord Drug Targets. 2017;17:100–113.
- Resnick ES, Moshier EL, Godbold JH, et al. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650–1657.
- Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–286.
- Gathmann B, Mahlaoui N, Gerard L, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134:116–126.
- Khan R, Habbal M, Scaffidi MA, et al. Gastrointestinal disease in patients with common variable immunodeficiency: a retrospective observational study. J Can Assoc Gastroenterol. 2020;3(4):162–168.
- Ho H-E, Cunningham-Rundles C. Non-infectious complications of common variable immunodeficiency: updated clinical spectrum, sequelae, and insights to pathogenesis. Front Immunol. 2020;11. DOI:https://doi.org/10.3389/fimmu.2020.00011.
- Takiishi T, Fenero CIM, Câmara NOS., et al. Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers. 2017;5:e1373208.
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48.
- Pikkarainen S, Martelius T, Ristimaki A, et al. A high prevalence of gastrointestinal manifestations in common variable immunodeficiency. Am J Gastroenterol. 2019;114:648–655.
- Farmer JR, Ong M-S, Barmettler S, et al. Common variable immunodeficiency non-infectious disease endotypes redefined using unbiased network clustering in large electronic datasets. Front Immunol. 2018;8:1740.
- Hermans PE, Diaz-Buxo JA, Stobo JD., et al. Idiopathic late-onset immunoglobulin deficiency: clinical observations in 50 patients. Am J Med. 1976;61(2):221–237.
- Hermaszewski R, Webster A. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. QJM. 1993;86:31–42.
- Washington K, Stenzel TT, Buckley RH, et al. Gastrointestinal pathology in patients with common variable immunodeficiency and X-linked agammaglobulinemia. Am J Surg Pathol. 1996;20:1240–1252.
- Jorgensen SF, Reims HM, Frydenlund D, et al. A cross-Sectional study of the prevalence of gastrointestinal symptoms and pathology in patients with common variable immunodeficiency. Am J Gastroenterol. 2016;111:1467–1475.
- Mannon PJ, Fuss IJ, Dill S, et al. Excess IL-12 but not IL-23 accompanies the inflammatory bowel disease associated with common variable immunodeficiency. Gastroenterology. 2006;131(3):748–756.
- Sifers T, Hirten R, Mehandru S, et al. Vedolizumab therapy in common variable immune deficiency associated enteropathy: a case series. Clin Immunol. 2020;212:108362.
- Pecoraro A, Nappi L, and Crescenzi L, et al. Chronic diarrhea in common variable immunodeficiency: a case series and review of the literature. J Clin Immunol. 2018; 38: 67–76.
- Woodward JM, Gkrania-Klotsas E, Cordero-Ng AY, et al. The role of chronic norovirus infection in the enteropathy associated with common variable immunodeficiency. Am J Gastroenterol. 2015;110:320–327.
- Biagi F, Bianchi PI, Zilli A, et al. The significance of duodenal mucosal atrophy in patients with common variable immunodeficiency: a clinical and histopathologic study. Am J Clin Pathol. 2012;138:185–189.
- Rolfes MC, Sriaroon P, Dávila Saldaña BJ, et al. Chronic norovirus infection in primary immune deficiency disorders: an international case series. Diagn Microbiol Infect Dis. 2019;93:69–73.
- Jain P, Mishra A, and Gupta D, et al. Chronic enteropathy-related malabsorption syndrome in an adult with common variable immunodeficiency and symptomatic norovirus infection of the gut. BMJ Case Rep. 2021;14:1–4.
- Duraisingham S, Manson A, Grigoriadou S, et al. Immune deficiency: changing spectrum of pathogens. Clin Exp Immunol. 2015;181:267–274.
- Van De Ven A, Douma JW, and Rademaker C, et al. Case report-Pleconaril-resistant chronic parechovirus-associated enteropathy in agammaglobulinaemia. Antivir Ther. 2011;16:611.
- Radanović I, Rkman D, Zekan P, et al. Chronic meningoencephalitis caused by echo virus 6 in a patient with common variable immunodeficiency: successful treatment with pleconaril. Wien Klin Wochenschr. 2018;130:70–72.
- Maarschalk-Ellerbroek LJ, Oldenburg B, Mombers IM, et al. Outcome of screening endoscopy in common variable immunodeficiency disorder and X-linked agammaglobulinemia. Endoscopy. 2013;45(4):320–323.
- Malamut G, VerkarreV, Suarez F, et al. The enteropathy associated with common variable immunodeficiency: the delineated frontiers with celiac disease. Am J Gastroenterol. 2010; 105(10): 2262–2275.
- van der Poorten DK, McLeod D, and Ahlenstiel G, et al. Gastric cancer screening in common variable immunodeficiency. J Clin Immunol. 2018;38:768–777.
- Maccaferri S, Vitali B, Klinder A, et al. Rifaximin modulates the colonic microbiota of patients with crohn’s disease: an in vitro approach using a continuous culture colonic model system. J Antimicrob Chemother. 2010;65:2556–2565.
- Zullo A, Romiti A, Rinaldi V, et al. Gastric pathology in patients with common variable immunodeficiency. Gut. 1999;45(1):77–81.
- Vajdic CM, Mao L, van Leeuwen MT, et al. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood. 2010;116(8):1228–1234.
- Venhoff N, Emmerich F, and Neagu M, et al. The role of HLA DQ2 and DQ8 in dissecting celiac-like disease in common variable immunodeficiency. J Clin Immunol. 2013;33:909–916.
- Daniels JA, Lederman HM, Maitra A, et al. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): a clinicopathologic study and review. Am J Surg Pathol. 2007;31:1800–1812.
- Luzi G, Zullo A, Iebba F, et al. Duodenal pathology and clinical-immunological implications in common variable immunodeficiency patients. Am J Gastroenterol. 2003;98:118–121.
- Pita JS, Fernandes RAR, and Almeida R, et al. Gluten-free diet: a possible treatment for chronic diarrhoea in common variable immunodeficiency. BMJ Case Rep. 2018:.
- Biesiekierski JR, Muir JG, Gibson PR., et al. Is gluten a cause of gastrointestinal symptoms in people without celiac disease? Curr Allergy Asthma Rep. 2013;13:631–638.
- Jorgensen SF, Reims HM, Aukrust P, et al. CVID and celiac disease. Am J Gastroenterol. 2017;112:393.
- Skodje GI, Minelle IH, Rolfsen KL, et al. Dietary and symptom assessment in adults with self-reported non-coeliac gluten sensitivity. Clin Nutr ESPEN. 2019;31:88–94.
- Aslam A, Misbah SA, Talbot K, et al. Vitamin E deficiency induced neurological disease in common variable immunodeficiency: two cases and a review of the literature of vitamin E deficiency. Clin Immunol. 2004;112:24–29.
- Agarwal S, Mayer L. Diagnosis and treatment of gastrointestinal disorders in patients with primary immunodeficiency. Clin Gastroenterol Hepatol. 2013;11:1050–1063.
- Byrne MF, Royston D, and Patchett SE., et al. Association of common variable immunodeficiency with atypical collagenous colitis. Eur J Gastroenterol Hepatol. 2003;15:1051–1053.
- Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Q J Med. 1993;86:31–42.
- Khodadad A, Aghamohammadi A, Parvaneh N, et al. Gastrointestinal manifestations in patients with common variable immunodeficiency. Digestive Dis Sci. 2007;52:2977–2983.
- Wood P, Stanworth S, Burton J, et al. Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systematic review. Clin Exp Immunol. 2007;149:410–423.
- Teahon K, Webster AD, Price AB, et al. Studies on the enteropathy associated with primary hypogammaglobulinaemia. Gut. 1994;35:1244–1249.
- Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380:1590–1605.
- Boursiquot JN, Gerard L, and Malphettes M, et al. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol. 2013;33:84–95.
- Ö A, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol. 2009;133:198–207.
- Slade CA, Bosco JJ, Binh Giang T, et al. Delayed diagnosis and complications of predominantly antibody deficiencies in a cohort of Australian adults. Front Immunol. 2018;9:694.
- Azzu V, Fonseca M, Duckworth A, et al. Liver disease is common in patients with common variable immunodeficiency and predicts mortality in the presence of cirrhosis or portal hypertension. J Allergy Clin Immunol Pract. 2019;7(7):2484–6 e3.
- Pecoraro A, Crescenzi L, Varricchi G, et al. Heterogeneity of liver disease in common variable immunodeficiency disorders. Front Immunol. 2020;11:338.
- Ziol M, Poirel H, Kountchou GN, et al. Intrasinusoidal cytotoxic CD8+ T cells in nodular regenerative hyperplasia of the liver. Hum Pathol. 2004;35:1241–1251.
- Malamut G, Ziol M, Suarez F, et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol. 2008;48(1):74–82.
- Fuss IJ, Friend J, and Yang Z, et al. Nodular regenerative hyperplasia in common variable immunodeficiency. J Clin Immunol. 2013;33:748–758.
- Reshamwala PA, Kleiner DE, Heller T., et al. Nodular regenerative hyperplasia: not all nodules are created equal. Hepatology. 2006;44(1):7–14.
- Ward C, Lucas M, Piris J, et al. Abnormal liver function in common variable immunodeficiency disorders due to nodular regenerative hyperplasia. Clin Exp Immunol. 2008;153(3):331–337.
- Jorgensen SF, Macpherson ME, Bjoro K, et al. Liver transplantation in patients with primary antibody deficiency. J Allergy Clin Immunol. 2017;139:1708–10 e2.
- Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology. 1990;11:787–797.
- Colina F, Alberti N, Solis JA, et al. Diffuse nodular regenerative hyperplasia of the liver (DNRH). A clinicopathologic study of 24 cases. Liver. 1989;9:253–265.
- Perez Ruiz F, Orte Martinez FJ, Zea Mendoza AC, et al. Nodular regenerative hyperplasia of the liver in rheumatic diseases: report of seven cases and review of the literature. Semin Arthritis Rheum. 1991;21:47–54.
- Xiao X, Miao Q, Chang C, et al. Common variable immunodeficiency and autoimmunity–an inconvenient truth. Autoimmun Rev. 2014;13:858–864.
- Azizi G, Abolhassani H, Asgardoon MH, et al. Autoimmunity in common variable immunodeficiency: epidemiology, pathophysiology and management. Expert Rev Clin Immunol. 2017;13:101–115.
- Mahdavinia M, Mirsaeidi M, Bishehsari F, et al. Primary sclerosing cholangitis in common variable immune deficiency. Allergol Int. 2015;64:187–189.
- Apostolov R, Sinclair M, and Lokan J, et al. Successful liver transplantation in common variable immune deficiency with reversal of hepatopulmonary syndrome. BMJ Case Rep. 2019;12:1–3.
- Azzu V, Elias JE, Duckworth A, et al. Liver transplantation in adults with liver disease due to common variable immunodeficiency leads to early recurrent disease and poor outcome. Liver Transpl. 2018;24:171–181.
- Montalti R, Mocchegiani F, and Vincenzi P, et al. Liver transplantation in patients with common variable immunodeficiency: a report of two cases. Ann Transplant. 2014; 19: 541–544.
- Murakawa Y, Miyagawa-Hayashino A, Ogura Y, et al. Liver transplantation for severe hepatitis in patients with common variable immunodeficiency. Pediatr Transplant. 2012;16:E210–6.
- Chen Y, Cameron A. Aspergillosis after liver transplantation in the context of common variable immunodeficiency: case report. Transpl Infect Dis. 2013;15:540–544.
- Smith MS, Webster AD, Dhillon AP, et al. Orthotopic liver transplantation for chronic hepatitis in two patients with common variable immunodeficiency. Gastroenterology. 1995;108:879–884.
- Hadzic N, Pagliuca A, Rela M, et al. Correction of the hyper-IgM syndrome after liver and bone marrow transplantation. N Engl J Med. 2000;342:320–324.
- Gow PJ, Mutimer D. Successful outcome of liver transplantation in a patient with hepatitis C and common variable immune deficiency. Transpl Int. 2002;15:380–383.
- Rodrigues F, Davies EG, Harrison P, et al. Liver disease in children with primary immunodeficiencies. J Pediatr. 2004;145:333–339.
- Emerson J, van der Poorten DK, Lin MW, et al. Duodenal plasma cells correspond to serum IgA in common variable immunodeficiency. Pathology. 2020. DOI:https://doi.org/10.1016/j.pathol.2020.08.014.
- Agarwal S, Mayer L. Gastrointestinal manifestations in primary immune disorders. Inflamm Bowel Dis. 2010;16:703–711.
- Arieira C, Dias de Castro F, Moreira MJ, et al. Common variable immunodeficiency-Associated inflammatory enteropathy: the new era of biological therapy. GE Port J Gastroenterol. 2018;25:322–326.
- Chua I, Standish R, Lear S, et al. Anti-tumour necrosis factor-alpha therapy for severe enteropathy in patients with common variable immunodeficiency (CVID. Clin Exp Immunol. 2007;150:306–311.
- Vazquez-Moron JM, Pallares-Manrique H, Martin-Suarez IJ, et al. Crohn’s-like disease in a patient with common variable immunodeficiency treated with azathioprine and adalimumab. Rev Esp Enferm Dig. 2013;105:299–302.
- Boland BS, Riedl MA, Valasek MA, et al. Vedolizumab in patients with common variable immune deficiency and gut inflammation. Am J Gastroenterol. 2017;112:1621.
- Akhtar HJ, Markandey B, Ma C, et al. Vedolizumab induced clinical, endoscopic, and histological improvement in common variable immunodeficiency disease-associated intestinal enteropathy. Inflamm Bowel Dis. 2020;26:e22–e3.
- Navarini AA, Hruz P, Berger CT, et al. Vedolizumab as a successful treatment of CTLA-4-associated autoimmune enterocolitis. J Allergy Clin Immunol. 2017;139:1043–6.e5.
- Egg D, Rump IC, Mitsuiki N, et al. Therapeutic options for CTLA-4 insufficiency. J Allergy Clin Immunol. 2021. DOI:https://doi.org/10.1016/j.jaci.2021.04.039.
- Kiaee F, Azizi G, Rafiemanesh H, et al. Malignancy in common variable immunodeficiency: a systematic review and meta-analysis. Expert Rev Clin Immunol. 2019;15:1105–1113.
- Pulvirenti F, Pecoraro A, Cinetto F, et al. Gastric cancer is the leading cause of death in Italian adult patients with common variable immunodeficiency. Front Immunol. 2018;9:2546.
- Correa P. Human gastric carcinogenesis: a multistep and multifactorial process–First American cancer society award lecture on cancer epidemiology and prevention. Cancer Res. 1992;52:6735–6740.
- Rawla P, Barsouk A. Epidemiology of gastric cancer: global trends, risk factors and prevention. Prz Gastroenterol. 2019;14:26.
- De Petris G, Dhungel BM, and Chen L, et al. Gastric adenocarcinoma in common variable immunodeficiency:features of cancer and associated gastritis may be characteristic of the condition. Int J Surg Pathol. 2014; 22: 600–606.
- Gullo I, Costa C, and Silva SL, et al. The dysfunctional immune system in common variable immunodeficiency increases the susceptibility to gastric cancer. Cells. 2020;9.
- Thaventhiran JED, Lango Allen H, and Burren OS, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. 2020;583(7814):90–95 .
- O’Reilly LA, Putoczki TL, Mielke LA, et al. Loss of NF-κB1 causes gastric cancer with aberrant inflammation and expression of immune checkpoint regulators in a STAT-1-Dependent manner. Immunity. 2018;48:570–83.e8.
- Jorgensen SF, Fevang B, and Aukrust P., et al. Autoimmunity and inflammation in CVID: a possible crosstalk between immune activation, gut microbiota, and epigenetic modifications. J Clin Immunol. 2019; 39: 30–36.
- Desar IM, Keuter M, and Raemaekers JM, et al. Extranodal marginal zone (MALT) lymphoma in common variable immunodeficiency. Neth J Med. 2006;64: 136–140.
- Larvol L, Cervoni JP, and Hagiage M, et al. [Colonic lymphoma simulating cryptogenetic colitis associated with common variable hypogammaglobulinemia]. Gastroenterol Clin Biol. 1994;18: 779–781.
- Wehr C, Houet L, and Unger S, et al. Altered spectrum of lymphoid neoplasms in a single-Center cohort of common variable immunodeficiency with immune dysregulation. J Clin Immunol. 2021. DOI:https://doi.org/10.1007/s10875-021-01016-4.
- Khalil MO, Morton LM, Devesa SS, et al. Incidence of marginal zone lymphoma in the United States, 2001–2009 with a focus on primary anatomic site. Br J Haematol. 2014;165:67–77.
- Murri M, Leiva I, Gomez-Zumaquero JM, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 2013;11(1):1–12.
- Miyake S, Kim S, Suda W, et al. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS One. 2015;10(9):e0137429.
- Zhang X, Zhang D, Jia H, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 2015;21:895–905.
- Jorgensen SF, Troseid M, Kummen M, et al. Altered gut microbiota profile in common variable immunodeficiency associates with levels of lipopolysaccharide and markers of systemic immune activation. Mucosal Immunol. 2016; 9(6): 1455–1465.
- Fadlallah J, El Kafsi H, Sterlin D, et al. Microbial ecology perturbation in human IgA deficiency. Sci Transl Med. 2018;10:eaan1217.
- Shulzhenko N, Dong X, Vyshenska D, et al. CVID enteropathy is characterized by exceeding low mucosal iga levels and interferon-driven inflammation possibly related to the presence of a pathobiont. Clin Immunol. 2018. DOI:https://doi.org/10.1016/j.clim.2018.09.008.
- Fadlallah J, Sterlin D, Fieschi C, et al. Synergistic convergence of microbiota-specific systemic IgG and secretory IgA. J Allergy Clin Immunol. 2019;143:1575–85 e4.
- Fiedorová K, Radvanský M, Bosák J, et al. Bacterial but not fungal gut microbiota alterations are associated with common variable immunodeficiency (CVID) phenotype. Front Immunol. 2019;10:1914.
- Franco-Esquivias AP, Peña CG, and Torres-Lozano C, et al. Gut microbiota in Mexican patients with common variable immunodeficiency. Gac Med Mex. 2019;155:447–452.
- Jorgensen SF, Holm K, Macpherson ME, et al. Selective IgA deficiency in humans is associated with reduced gut microbial diversity. J Allergy Clin Immunol. 2019;143:1969–71 e11.
- Jorgensen SF, Macpherson ME, Bjornetro T, et al. Rifaximin alters gut microbiota profile, but does not affect systemic inflammation - a randomized controlled trial in common variable immunodeficiency. Sci Rep. 2019;9:167.
- van Schewick CM, Nöltner C, Abel S, et al. Altered microbiota, impaired quality of life, malabsorption, infection, and inflammation in CVID patients with diarrhoea. Front Immunol. 2020;11:1654.
- Bosák J, Lexa M, Fiedorová K, et al. Patients with common variable immunodeficiency (CVID) show higher gut bacterial diversity and levels of low-Abundance genes than the healthy housemates. Front Immunol. 2021;12:671239.
- Baniadam L, Arshi S, and Nabavi M, et al. Can concurrent lower gastrointestinal manifestations help the timely diagnosis of small intestinal bacterial overgrowth in CVID patients? Eur Ann Allergy Clin Immunol. 2021; 53: 18–22.
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11:373–384.
- Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249.
- Macpherson ME, Hov JR, Ueland T, et al. Gut microbiota-Dependent Trimethylamine N-Oxide associates with inflammation in common variable immunodeficiency. Front Immunol. 2020;11:574500.
- Trøseid M, Ueland T, Hov JR, et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med. 2015;277:717–726.
- Missailidis C, Neogi U, Stenvinkel P, et al. The microbial metabolite trimethylamine-N-oxide in association with inflammation and microbial dysregulation in three HIV cohorts at various disease stages. AIDS. 2018;32(12):1589–1598.
- Janeiro MH, Ramirez MJ, and Milagro FI, et al. Implication of Trimethylamine N-Oxide (TMAO) in disease: potential biomarker or new therapeutic target. Nutrients. 2018;10:1–22.
- Sun X, Jiao X, Ma Y, et al. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem Biophys Res Commun. 2016;481:63–70.
- Chen ML, Zhu XH, and Ran L, et al. Trimethylamine-N-Oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. 2017;6(9):1–17.
- Mohammed AD, Khan MAW, Chatzistamou I, et al. Gut antibody deficiency in a mouse model of CVID results in spontaneous development of a gluten-Sensitive enteropathy. Front Immunol. 2019: 10. DOI:https://doi.org/10.3389/fimmu.2019.00010.
- Jørgensen SF, Fevang B, Aukrust P., et al. Commentary: gut antibody deficiency in a mouse model of CVID results in spontaneous development of a gluten-Sensitive enteropathy. Front Immunol. 2020;11:1921.
- Arieira C, Dias de Castro F, and Moreira MJ, et al. Common variable immunodeficiency-Associated inflammatory enteropathy: the new era of biological therapy. GE Port J Gastroenterol. 2018;25(6):322–326.
- Geuking MB, Cahenzli J, Lawson MA, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806.
- Hooper LV, Littman DR, Macpherson AJ., et al. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–1273.
- Sutherland DB, Fagarasan S. IgA synthesis: a form of functional immune adaptation extending beyond gut. Curr Opin Immunol. 2012;24:261–268.
- Lorenz RG, Chaplin DD, McDonald KG, et al. Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin beta receptor, and TNF receptor I function. J Immunol. 2003;170:5475–5482.
- Kaetzel CS. Cooperativity among secretory IgA, the polymeric immunoglobulin receptor, and the gut microbiota promotes host-microbial mutualism. Immunol Lett. 2014;162:10–21.
- Li J, Jorgensen SF, Maggadottir SM, et al. Association of CLEC16A with human common variable immunodeficiency disorder and role in murine B cells. Nat Commun. 2015;6(1):6804.
- Hirschfield GM, Xie G, Lu E, et al. Association of primary biliary cirrhosis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun. 2012;13(4):328–335.
- Mells GF, Floyd JA, Morley KI, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43:329–332.
- Dubois PC, Trynka G, Franke L, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302.
- Márquez A, Varadé J, Robledo G, et al. Specific association of a CLEC16A/KIAA0350 polymorphism with NOD2/CARD15(-) crohn’s disease patients. Eur J Hum Genet. 2009;17:1304–1308.
- Mardinoglu A, Wu H, Bjornson E, et al. An integrated understanding of the rapid metabolic benefits of a Carbohydrate-Restricted diet on hepatic steatosis in humans. Cell Metab. 2018;27(3):559–71 e5.
- Consortium HMP. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214.
- Zeevi D, Korem T, Zmora N, et al. Personalized nutrition by prediction of glycemic responses. Cell. 2015;163(5):1079–1094.
- Mendes-Soares H, Raveh-Sadka T, Azulay S, et al. Assessment of a personalized approach to predicting postprandial glycemic responses to food among individuals without diabetes. JAMA Network Open. 2019;2(2):e188102.