
1. Introduction
Tumor necrosis factor (TNF) and TNF-receptor (TNFR) superfamilies of proteins (TNFSF and TNFRSF, respectively) consist of several molecules with critical participation in a vast array of biological processes, including a pivotal role in the development and function of the immune system. TNF-like cytokine 1A (TL1A) that is encoded by the TNFSF15 gene was first described in 2002 [Citation1]. The product is a type II transmembrane protein, which may exist in either membrane-bound or soluble forms, which are both functional but may exert diverse immunological effects. TL1A signals via binding to its functional receptor, death domain receptor 3 (DR3 or TNFFRSFf25), which shares the highest homology to TNFR1 among all members of the TNFRSF [Citation2]. During the two decades that followed its original report, the TL1A/DR3 system of cytokines has arisen as an important module for mucosal immunity that is involved in the preservation of intestinal homeostasis but also critically participates in the development and maintenance of chronic inflammatory responses that take place in patients with inflammatory bowel diseases (IBD) [Citation2].
2. The immunological basis for targeting TL1A in IBD
2.1. TL1A/DR3 signaling induces pro-inflammatory responses
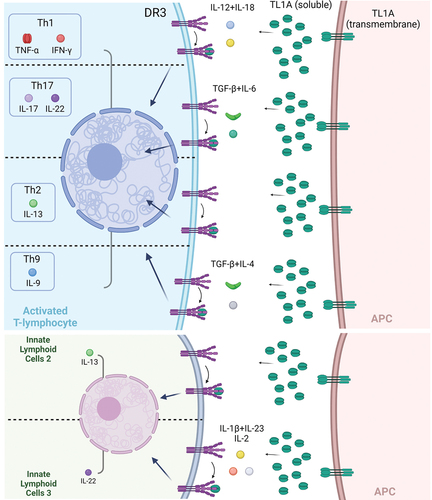
Although TL1A was originally described as an endothelial factor, its cellular distribution has, since, expanded to include mainly antigen-presenting cells (APCs) (dendritic cells, macrophages), but also lymphocytes, plasma cells, and fibroblasts. On the other hand, the expression of DR3 is largely confined to activated lymphocytes. This strategic positioning of TL1A and DR3 in APCs and lymphocytes, respectively, allows them to act as powerful co-stimulators of immunological responses with enhanced specificity for mucosal tissues (). Indeed, in various experimental and clinical systems, the binding of TL1A on DR3-expressing lymphocytes induces stimulatory signals which are capable of amplifying all major effector T-cell responses, including Th1, Th17, Th2 and Th9. Lymphocytes from mucosal sites are particularly sensitive to TL1A stimulation, including specific subpopulations that are signified by the expression of CCR9, CD161, and IL-18Rα and participate in the pathogenesis of IBD-like chronic intestinal inflammation [Citation3]. Accordingly, downstream effects of TL1A/DR3 signaling include the production of several pivotal pro-inflammatory cytokines such as IFN-γ, TNF-α, IL-6 and IL-17, as well as IL-2, IL-4, IL-13, all of which are upregulated in clusters of patients with IBD and may bare pathogenetic potential. DR3 is also highly expressed by innate lymphoid cells (ILCS), in particular ILC2 (and ILC3), which respond to stimulation by TL1A with aberrant cytokine secretion [Citation4].
Figure 1. The multifaceted function of TL1A/DR3 signaling in mucosal immunity. TL1A is primarily produced by Antigen Presenting Cells (APCs) and can be expressed as a transmembrane protein or be cleaved and secreted in a soluble form. Both configurations are capable of binding to the functional receptor death domain receptor 3 which is mainly expressed by activated lymphocytes and innate lymphoid cells (ILC). TL1A/DR3 acts as a co-stimulatory module, the final effect of which is affected by the cytokine milieu of the specific tissue microenvironment and can be Th1, Th2, Th17, Th9 predominant but can also affect regulatory lymphocytes and ILC2 and ILC3 function. Consequently, TL1A blockade may be proven of therapeutic value for a variety of chronic inflammatory diseases that may be dominated by diverse T-effector immunophenotypes. Created with BioRender.com.
2.2. Overexpression of TL1A/DR3 leads to mucosal inflammation
Independent research groups have generated transgenic mice that overexpress TL1A exclusively to lymphoid or myeloid cells, respectively. All murine strains developed intestinal inflammation that was localized at the ileum and was characterized by epithelial changes, inflammatory infiltration of the lamina propria and thickening of the muscularis layer [Citation5]. Ileitis was characterized immunologically by increases in activated T cells, predominantly of the Th2-type and regulatory Foxp3+ CD4 + T-cells. Although the severity of intestinal inflammation in TL1A-Tg mice was mild and confined to the small intestine, their response to acute environmental injury was highly compromised. In fact, upon challenge with dextran sulfate sodium (DSS), TL1A-transgenic mice developed more severe colitis in comparison to wild-type controls. This was probably mediated via aberrant activation and migration of dendritic cells, which showed increased phagocytotic and antigen-presenting ability and overexpressed inflammatory cytokines (TNF-α and IL-12/23 p40) and chemokine receptors (CCR5 and CCR7) [Citation6]. The proinflammatory role of the TL1A/DR3 system was also shown recently in the SAMP1/YitFc, ileitis-prone mouse, since the administration of an agonistic anti-DR3 antibody worsened ileitis, in association with upregulation of mucosal Th1 and Th2 cytokines, expansion of dysfunctional CD25-FoxP3+ cells and increased mucosal ratio of ILC1:ILC3 [Citation7]. Finally, administration of exogenous TL1A highly exacerbated cytokine (IL-12 plus IL-18)-induced colitis and ileitis in a murine model, in association with epithelial damage, and increased colonic levels of inflammatory mediators [Citation8].
2.3. Silencing of TL1A/DR3 signaling has anti-inflammatory effects in preclinical studies
Several studies have reported the effects of genetic deletion or pharmaceutical blockade of the TL1A/DR3 system on the severity of experimental intestinal inflammation. DR3 and TL1Ako mice do not develop pathologies, spontaneously, but are protected from Th17 pathology, possibly due to an inability of TL1A-deficient dendritic cells to support the differentiation and proliferation of Th17 lymphocytes [Citation9]. In the aforementioned SAMP1/YitFc mouse model of CD, genetic deletion of DR3 effectively reversed the inflammatory phenotype and induced selective expansion of CD25+ FoxP3+ over CD25-FoxP3+ cells and upregulation of IL-10. Furthermore, the effects of TL1A/DR3 blockade were also investigated in several murine models of chronic colitis, including the chronic-DSS, the 2,4,6-trinitrobenzosulfonic acid (TNBS), and the G-protein ai2 deficient models. In all cases, a significant local upregulation of TL1A and DR3 coincided with the development of large intestinal inflammation. The pathogenetic significance of such elevations was demonstrated by the amelioration of the severity of colitis after treatment with anti-TL1A monoclonal antibodies. Collectively, these data point to a critical role of TL1A/DR3 responses in the finetuning of mucosal immunity during chronic inflammation, including the regulation of the relative abundance of T effectors vs. Tregs and raise the case for applying anti-TL1A therapies in patients with IBD.
3. The clinical basis for targeting TL1A/DR3 in IBD
3.1. TL1A/DR3 are upregulated in inflamed mucosal tissues from patients with IBD
Extensive characterization of mucosal expression profiles has clearly demonstrated increased expression of TL1A and DR3 in areas with active inflammation both at the mRNA and protein levels, in patients with either ulcerative colitis (UC) or Crohn’s disease (CD) [Citation10]. Immunolocalization of TL1A was reported in tissue macrophages, lymphocytes, and infiltrating plasma cells, which was significantly increased in inflamed tissues from patients with IBD and correlated with the intensity of inflammatory changes. The expression of DR3 was also increased and localized exclusively in T-lymphocytes. Further characterization of mucosal TL1A-producing mucosal cells identified a population of relevant lamina propria mononuclear phagocytes which displayed markers of antigen-sampling cells, as was indicated by the expression of CD14 and CX3CR1. A positive correlation was also reported between TL1A production by this population and disease activity in patients with CD [Citation11].
3.2. Polymorphisms in the TNFSF15 gene modify the risk for developing IBD
Initial studies in patients of different ethnic backgrounds showed the existence of 3 haplotypes for TNFSF15 that confer either increased risk or protection from developing IBD. Those associations were validated in additional patient cohorts and are considered among the most prevalent around the globe, although TNFSF15 variants appear to have a stronger association with IBD susceptibility in Asian populations [Citation12]. Two additional observations are also worth mentioning. Firstly, haplotype-phenotype associations for TNFSFf15 polymorphisms have been proposed, as haplotype B predicted higher production of TL1A protein in response to FcγR or LPS stimulation. Secondly, TNFSF15 alleles may bear prognostic value for the severity of IBD. Indeed, in various studies certain haplotypes have been shown to be associated with stricturing, penetrating, or perianal complications, disease progression from B1 to B2/B3 Montreal phenotypes, or medically refractory UC [Citation13]. Finally, an association between specific TNFSF15 alleles and microbiome composition was also shown.
3.3. TL1A blockade has shown promising results in early clinical drug development studies
The aforementioned clinical and experimental evidence encouraged the development of a fully human, IgG1 anti-TL1A mAb [PF-06480605] for use in patients. There are two officially registered clinical trials with this mAb in patients with Ulcerative colitis, and their results have been published [Citation14,Citation15]. In the phase 1 study, the pharmacokinetics, safety and immunogenicity of PF-06480605 were studied. The drug was well tolerated in the maximum studied doses both for single (800 mg x 1 iv) and multiple (500 mg x 3 iv) ascending regimens, without any serious adverse events or increased treatment-related adverse events in comparison to the placebo group. This prompted the subsequent phase 2a TUSCANY study, whereby 50 patients with moderate to severe UC received 7 bi-weekly iv infusions of 500 mg of PF-06480605. The antibody was again well tolerated, with 3 patients experiencing SAE and two discontinuing treatments. The primary efficacy endpoint of endoscopic improvement was met by 38% of patients, with 24% achieving clinical remission and 10% endoscopic remission 14 weeks after drug initiation. In addition, a significant proportion demonstrated minimal histologic disease. Although a placebo arm was not included in the study, those results are numerically comparable to the efficacy indicators for clinically available biologics for UC. In further analysis, the investigators examined tissue, blood and fecal samples of patients and were able to dissect the mucosal and systemic immunological effects of anti-TL1A blockade [Citation16]. In particular, they demonstrated a significant post-treatment reduction of tissue TL1A with a simultaneous increase of serum TL1A, which was attributed to the effective binding of PF-06480605 with TL1A. Adding to this, patients with endoscopic improvement had higher pretreatment tissue TL1A levels, indicating that it could serve as a promising predictive biomarker. Moreover, they demonstrated tissue treatment-specific reductions in certain inflammatory populations of Th-17 cells and macrophages, a finding that was further supported by proteomic analysis in the peripheral blood. In addition, fibrotic pathways were also significantly affected by anti-TL1A treatment. Interestingly, analysis of pre- and post-PF-06480605 treatment fecal samples revealed a significant reduction of IBD-associated pathobionts.
4. Future perspectives
Based on the encouraging initial results, clinical programs for the therapeutic use of TL1A neutralization in IBD, most probably, will continue to develop. The final outcome of such an effort, eventually, will depend upon several immunological and clinical issues. Regarding the former, it should be remembered that, although the pro-inflammatory effects of TL1A/DR3 in chronic inflammation are indisputable, these proteins are involved in homeostatic mechanisms, also. TL1A- and DR3-deficient mice suffer from more severe acute-DSS colitis than wild-type controls, emphasizing their protective properties during acute injury and repair [Citation17]. It is worth noting that exacerbation of UC was the most common adverse event in the phase 2 trial of PF-06480605 [Citation15]. In addition, DR3 is expressed in all lineages of effector T-cells but also by Tregs and ILC2s. Thus, TL1A/DR3 signaling lacks specificity and may affect both pro- and anti-inflammatory pathways. Such dichotomy may prove critical in dictating the final outcome of TL1A inhibition. From the clinical standpoint, issues of concern include the inconvenient administration regimen of repeated iv infusions which will require frequent hospital visits, and the high immunogenicity rates that were reported. In particular, 82% of patients participating in the clinical program of PF-06480605 tested positive for anti-drug antibodies, with 10% developing neutralizing antibodies [Citation15]. Furthermore, the long-term effect of TL1A inhibition is not currently known due to the lack of a maintenance clinical trial at the moment.
Those concerns notwithstanding, several positive perspectives of anti-TL1A therapies should also be emphasized. First, TL1A was shown to be upregulated not only in association with intestinal but also joint [Citation18] and skin inflammation [Citation19], data that place this cytokine at the center of the gut-synovium-skin axis and increases its potential therapeutic applicability in IBD patients with extra-intestinal manifestations. Second, TL1A/DR3 signaling appears to be more potent in response to low-grade stimulation. This raises the possibility that anti-TLA treatment may be suitable for combinatorial therapies, leading to chronic control of low-grade inflammation after more potent treatments bring down the active disease. Third, several studies in a variety of chronic inflammatory have shown that soluble TL1A reliably correlates with active disease and responds to effective anti-inflammatory treatment, indicating that it may be used as a biomarker for disease activity and response to treatment. Finally, it was recently shown that TL1A/DR3 signaling is also involved in fibrogenesis. Human subepithelial intestinal myofibroblasts express TL1A in response to IBD-associated cytokines [Citation20], whereas elimination of TL1A leads to the prevention of intestinal strictures in mice [Citation21]. This renders anti-TL1A treatment a unique opportunity for the simultaneous targeting of inflammatory and pro-fibrotic pathways. In conclusion, targeting of TL1A/DR3 signaling may prove an efficient way to ameliorate intestinal and systemic inflammation in patients with IBD. Nevertheless, given the multiplicity of immunological effects of this cytokine/receptor system, further finetuning of its clinical application will be required. Single-cell transcriptomic analysis in combination with cellular immunophenotyping may facilitate the discovery of the relevant subpopulations of immunocytes that are more amenable to anti-TL1A blockade in terms of their modification in treatment responders. Another critical point would be the distinction between local and systemic effects of such blockade, inasmuch the former may provide modules with predictive value for response to treatment, whereas, the latter may be more helpful in the follow-up of the therapeutic effect. The role of DcR3, the decoy TL1A receptor, should also be explored in the future as this may provide an endogenous pathway for competing with DR3 for TL1A binding and prohibiting functional, pro-inflammatory signaling. Similarly, exploitation of the possibility for additional ligands for DR3 may also lead to therapeutic options. Finally, the role of TL1A during inflammation and fibrosis should be carefully dissected and inflammation-dependent mechanisms should be clearly discriminated from independent fibrogenesis, as this may set the background for fibrosis-reversing drug development.
Declaration of interest
G Bamias declares that he has acted as advisor/lecturer and/or received research funding and/or participates in clinical trials from AbbVie, Adacyte Therapeutics, Aenorasis, Amgen, Cooper, Ferring, Genesis Pharma, Janssen, MSD, Mylan, Pfizer, Takeda. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Migone TS, Zhang J, Luo X, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492.
- Valatas V, Kolios G, Bamias G. TL1A (TNFSF15) and DR3 (TNFRSF25): a co-stimulatory system of cytokines with diverse functions in gut mucosal immunity. Front Immunol. 2019;10. DOI:https://doi.org/10.3389/fimmu.2019.00583
- Jin S, Chin J, Seeber S, et al. TL1A/TNFSF15 directly induces proinflammatory cytokines, including TNFα, from CD3+CD161+ T cells to exacerbate gut inflammation. Mucosal Immunol. 2013;6:886–899.
- Meylan F, Hawley ET, Barron L, et al. The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol. 2014;7:958–968.
- Richard AC, Ferdinand JR, Meylan F, et al. The TNF-family cytokine TL1A: from lymphocyte costimulator to disease co-conspirator. J Leukoc Biol. 2015;98:333–345.
- Han F, Song J, Jia W, et al. TL1A primed dendritic cells activation exacerbated chronic murine colitis. Life Sci. 2020;262:118220.
- Li Z, Buttó LF, Buela KA, et al. Death receptor 3 signaling controls the balance between regulatory and effector lymphocytes in SAMP1/YitFc mice with crohn’s disease-like ileitis. Front Immunol. 2018;9:362.
- Tougaard P, Skov S, Pedersen AE, et al. TL1A regulates TCRγδ+ intraepithelial lymphocytes and gut microbial composition. Eur J Immunol. 2015;45:865–875.
- Pappu BP, Borodovsky A, Zheng TS, et al. TL1A–DR3 interaction regulates Th17 cell function and Th17-mediated autoimmune disease. J Exp Med. 2008;205:1049–1062.
- Bamias G, Martin C, Marini M, et al. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J Immunol. 2003;171:4868–4874.
- Castellanos JG, Woo V, Viladomiu M, et al. Microbiota-induced TNF-like Ligand 1A drives group 3 innate lymphoid cell-mediated barrier protection and intestinal T cell activation during colitis. Immunity. 2018;49:1077–1089.e5.
- Liu JZ, Van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–986.
- Ditrich F, Blümel S, Biedermann L, et al. Genetic risk factors predict disease progression in Crohn’s disease patients of the Swiss inflammatory bowel disease cohort. Therap Adv Gastroenterol. 2020;13:175628482095925.
- Banfield C, Rudin D, Bhattacharya I, et al. First-in-human, randomized dose-escalation study of the safety, tolerability, pharmacokinetics, pharmacodynamics and immunogenicity of PF-06480605 in healthy subjects. Br J Clin Pharmacol. 2020;86:812–824.
- Danese S, Klopocka M, Scherl EJ, et al. Anti-TL1A antibody PF-06480605 safety and efficacy for ulcerative colitis: a phase 2a single-arm study. Clin Gastroenterol Hepatol. 2021;19:2324–2332.e6.
- Hassan-Zahraee M, Ye Z, Xi L, et al. Antitumor necrosis factor-like ligand 1A therapy targets tissue inflammation and fibrosis pathways and reduces gut pathobionts in ulcerative colitis. Inflamm Bowel Dis. 2022;28:434–446.
- Jia L-G, Bamias G, Arseneau KO, et al. A novel role for TL1A/DR3 in protection against intestinal injury and infection. J Immunol. 2016;197(1):377–386.
- Konsta M, Bamias G, Tektonidou MG, et al. Increased levels of soluble TNF-like cytokine 1A in ankylosing spondylitis. Rheumatology. 2013;52(3):448–451.
- Li L, Fu L, Lu Y, et al. TNF-like ligand 1A is associated with the pathogenesis of psoriasis vulgaris and contributes to IL-17 production in PBMCs. Arch Dermatol Res. 2014;306(10):927–932.
- Bamias G, Filidou E, Goukos D, et al. Crohn’s disease-associated mucosal factors regulate the expression of TNF-like cytokine 1A and its receptors in primary subepithelial intestinal myofibroblasts and intestinal epithelial cells. Transl Res. 2017;180:118–130.e2.
- Shih DQ, Zheng L, Zhang X, et al. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol. 2014;7(6):1492–1503.