
ABSTRACT
Introduction
Several patients with myasthenia gravis (MG) do not adequately respond to available drugs or exhibit poor tolerance, necessitating the need for new therapies.
Areas covered
The paper discusses the rapidly evolving target-specific immunotherapies that promise long-standing remissions in the management of MG. It is specifically focused on the role of complement, anti-complement therapeutics, and the anti-FcRn and B cell monoclonals.
Expert opinion
Anti-AChR antibodies cause internalization of the receptors and activate complement leading to in situ MAC formation that damages the post-synaptic membrane of the neuromuscular junction. Inhibiting MAC formation by antibodies targeting key complements subcomponents is a reasonable therapeutic goal. Indeed, the anti-C5 monoclonal antibodies, Eculizumab, Ravulizumab, and Zilucoplan, have been successfully tested in MG with Eculizumab first and now Ravulizumab FDA-approved for refractory MG based on sustained long-term benefits. Among the biologics that inhibit FcRn, Efgartigimod caused rapid reduction of the circulating IgG in the lysosomes, and induced sustained clinical remission with good safety profile leading to FDA-approved indication. Anti-B cell agents, like Rituximab, can induce sustained long-term remissions, especially in IgG4 antibody-mediated Musk-MG, by targeting short-lived antibody-secreting plasmablasts. These biologics offer effective targeted immunotherapies with good tolerance promising to change the therapeutic algorithm in the chronic MG management.
1. Introduction
Myasthenia gravis (MG), being the prototypic antibody-mediated autoimmune disease where specific antibodies lead to endplate dysfunction, offers the opportunity for target-specific antibody therapies. Although we are doing well with nonspecific immunotherapies offering a gratifying response to at least 75% of the patients managing well the crises or the acute worsening with IVIg or plasmapheresis [Citation1–5], the chronic management for 25% of MG remains still challenging. At least 15% of MG patients can be refractory or incompletely responding to available therapies while in several cases long-term therapies with corticosteroid and immunosuppressants are either not well-tolerated or not routinely applied due to serious comorbidities. The rapidly evolving biologics are now powerful tools offering new options for long-standing clinical remissions with good tolerance and minimal long-term side effects [Citation1–5].
The review provides an update on three main categories of targeted biologics in the chronic management of generalized MG (g MG), centered on complement, FcRn and B cells, highlighting the promising results of just approved or soon to be likely approved new agents after successful Phase III clinical trials.
1.1. Clinical subtypes of MG in reference to current therapies and rationale for targeted immunotherapies
1.1.1. Clinical subtypes
The main Acetylcholine Receptor (AChR)-positive MG subtype that requires chronic targeted immunotherapies is the non-thymomatous g MG that comprises the Early-onset MG (EOMG), when the disease starts before the age of 50 and affects more women than men, the Late-onset MG (LOMG) when it starts above the age of 50 but below the age of 65, and the Very Late Onset (VLOMG) when starts above the age of 65 [Citation6]. Patients with LOMG and VLOMG are mostly men with ocular onset, while the VLOMG patients are often AChR-positive without thymoma and less frequently refractory to common drugs [Citation6]. Patients with g MG present with a varying degree of weakness in the bulbar, extraocular, facial and skeletal muscles manifested with dysphagia, diplopia and dysphonia, fatigable weakness in neck and proximal limb muscles, and in severe cases involvement of the respiratory muscles [Citation1,Citation4,Citation7]. Two major but distinct considerations that influence therapeutic decisions and the choice of medications relate to women with EOMG planning pregnancy and to patients with LOMG and VLOMG because of their frequent comorbidities [Citation6,Citation7]. An important subtype that also affects therapeutic decisions is the MG associated with Muscle-Specific-Kinase (Musk); these patients, comprising 6–8% of g MG, do not require thymectomy because their thymus lacks histological alterations and do not respond to anti-anticholinesterases [Citation8,Citation9]. Importantly, anti-Musk antibodies are of the IgG4 subclass and do not trigger antibody-mediated complement-fixing endplate destruction; as a result, the several anti-inflammatory agents such as IVIg or anti-complement therapeutics, are ineffective [Citation10–12] as discussed below. In non-thymomatous patients with g MG below the age of 50, therapeutic thymectomy is effective resulting in significantly reduced requirements of the maintenance steroid dose or immunosuppressants and decreased hospitalization rate [Citation13]; for LOMG patients above the age of 50, however, the results did not show significant benefit [Citation14]. Although the views are still divided, in our own practice, we do not recommend thymectomy in this age group.
1.1.2. Current therapies
g MG is a chronic disease requiring not only induction therapy with steroids but also maintenance with steroid-sparing immunosuppressants or immunomodulators. Therapy begins with (a) Anticholinesterases that offer mild symptomatic relief; (b) Corticosteroids that still remain the first-line drug, used by dose escalation from 20 mg daily to even up to 80–100 mg as needed, followed by slow tapering to every other day till reaching the lowest and safest effective dose that controls the disease; (c) Steroid-sparing Immunosuppressants such as Azathioprine, Mycophenolate Mofetil, Cyclosporine, or Tacrolimus, which have variable efficacy but also preference among practitioners, with most preferring mycophenolate 2,000 mg daily or Azathioprine (150 mg daily or 2–3 mg/kg) [Citation1–7]; and (d) Intravenous Immunoglobulin (IVIg) 2 g/kg over 3–4 days or plasmapheresis (every other day, six courses) for patients with significant worsening or MG crises [Citation1–5,Citation7,Citation15–18], choosing the one according to age, cost, availability, comorbidities and venous access [Citation1–5,Citation15–18]. IVIg is being also extensively used as maintenance therapy or as steroid-sparing agent, even though its long-term efficacy remains uncertain and has not been established with controlled studies, as discussed [Citation16]. Subcutaneous Immunoglobulin (sCIg) can be effective in reducing MG disability measures [Citation19], ensuring steady-state serum IgG levels for patients who exhibit wearing-off effect with IVIg or have poor venous access [Citation18].
1.1.3. Need for target-specific biologic therapies in MG
Despite the progress made with the above therapies and an overall increased survival and quality of life, there is still a need for better therapies collectively justified by the number of patients achieving remission based on disease burden, patient satisfaction, side effects, convenience, and quality of life assessments [Citation1–5]. New therapies would make a significant impact in treating g MG if they ensure long-term stability or remission, better tolerance, easy administration, and long-term safety. Such induction of benefits seems now promising by several biologic agents that target key molecules within the immunopathogenic network of MG considered responsible for the continuous immune activation and disease activity [Citation20].
1.2. Immunopathogenesis of MG as relate to target-specific therapies (Figure)
Although unclear what triggers MG, autoimmunity occurs when tolerance is broken and AChR proteins are presented to CD4 + T cells by Antigen-Presenting Cells, leading to upregulation of IL4 and IL6 cytokines which stimulate B cell to produce anti-AChR antibodies; these antibodies are pathogenic because they fix complement at the end-plate region leading to destruction of the AChRs and disruption of the neuromuscular transmission [Citation1,Citation3,Citation7,Citation21,Citation22] and . In addition to antigen-specific B cells, activated T cells secrete pro-inflammatory cytokines, like IFN-γ and IL-17, creating an imbalance between regulatory T-cells (Treg) and Th17-cells that further enhance antibody production [Citation1–3]. Targeted immunotherapies in MG as proposed several years ago [Citation20], have now become a clinical reality based on the impressive effectiveness of several biologic agents targeting B cells, Complement, and the neonatal Fc Receptor (FcRn) that enhances IgG antibody catabolism [Citation3] [ (*,**,***)]. The success of these agents, some of which have now gained FDA-approval for generalized MG, are slowly changing the therapeutic algorithm of the disease. The focus of this review is centered on these agents with the main emphasis on complement and anti-complement therapeutics.
Figure 1. Key biologic therapies in the immunopathogenic network of myasthenia gravis, targeting B cells (*), complement (**), and FcRn (***). The AChR, presented via APC’s to CD4 + T cells via co-stimulatory molecules lead to upregulation of cytokines that stimulate B cells to produce IgG anti-AChR antibodies. The AChR IgG by fixing complement at the end-plate region, leads to destruction of the AChR’s. treg and Th17+ cells, cytokines such as IL-6 that induce tregs, and proinflammatory cytokines, such as IL-17A, IL-21, and IL-22, sustain the immune imbalance. Promising biologic therapies are currently targeting B cells (*); complement (**), activated by the antibodies to cause destruction of AChR at the postsynaptic region via MAC formation; and FcRn (***), leading to increased catabolism of IgG-AChR antibodies reducing their pathogenic effects (adapted from Dalakas MC [Citation1–3].
![Figure 1. Key biologic therapies in the immunopathogenic network of myasthenia gravis, targeting B cells (*), complement (**), and FcRn (***). The AChR, presented via APC’s to CD4 + T cells via co-stimulatory molecules lead to upregulation of cytokines that stimulate B cells to produce IgG anti-AChR antibodies. The AChR IgG by fixing complement at the end-plate region, leads to destruction of the AChR’s. treg and Th17+ cells, cytokines such as IL-6 that induce tregs, and proinflammatory cytokines, such as IL-17A, IL-21, and IL-22, sustain the immune imbalance. Promising biologic therapies are currently targeting B cells (*); complement (**), activated by the antibodies to cause destruction of AChR at the postsynaptic region via MAC formation; and FcRn (***), leading to increased catabolism of IgG-AChR antibodies reducing their pathogenic effects (adapted from Dalakas MC [Citation1–3].](/cms/asset/ef4ce446-86fb-4c89-8886-1a1d73b30d2d/ierm_a_2082946_f0001_oc.jpg)
2. Complement and anti-complement biologics in the treatment of MG
2.1. Complement assembly
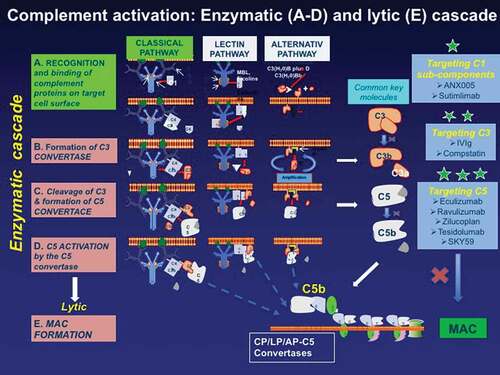
The complement system functionally consists of two main parts: the enzymatic cascade which generates the molecules needed to initiate the lytic pathway and the lytic pathway where the soluble proteins undergo conformational changes that lead to the formation of the C5b–91 membrane attack complex (MAC) [Citation11] (). The enzymatic cascade consists of the classical, the lectin, and the alternative pathways where a series of proteolytic enzyme complexes activate each other leading to the assembly of the convertases that drive subsequent complement activation and amplification, as detailed before [Citation23]. The classical pathway begins when immunoglobulin molecules of the IgG1-3 subclasses and of the IgM isotype (but not of the IgG4 subclass as the anti-Musk IgG antibodies are) bind to C1q altering its conformation; this in turn allows auto-activation of the C1r that cleaves the associated C1s in this complex finally enabling the formation of the surface-bound C5 convertase. The lectin pathway, activated by a wide array of carbohydrates, leads to the cleavage of C3 and formation of the C3 convertase, while the alternative pathway via a continuous activation process generates abundant C3a and C3b forming a C5 convertase, all finally leading to the production of C5a and C5b. The lytic pathway, after the cleavage of C5b to C5 convertase assembles the formation of MAC which forms osmolytic pores in the cell membranes ().
Figure 2. The main proteins involved in the complement activation cascade and the targeted complement therapeutics (highlighted with *). The complement begins with the enzymatic cascade that involves the assembly of enzyme complexes known as convertases. the enzyme cascade proceeds via three different activation pathways: the classical, the lectin, and the alternative pathway. the classical pathway begins with antibody-mediated activation of C1 that leads to the formation of the C4bC2a complex, which is the C3 convertase; the C3 convertase subsequently cleaves C3 to produce C3b, C4b C2a leading to C5 convertase complex that produces C5a and C5b. the lectin pathway begins with signal recognition by oligomeric structures which mediate the production of C4b proceeding thereafter as the classical pathway. In the alternative pathway, the C3 interacts through an amplification loop to form additional C3b that bind to C3-convertase forming a C5 convertase that cleaves C5. All three pathways generate C5b, which initiates the lytic pathway and the formation of membrane attack complex (MAC). The processes that represent therapeutic targets are shown on the the three boxes on the right along with drugs currently available or in development including: against C1* (ANX005 and Sutimilab); against C3** (IVIg and compstatin); and against C5*** (Eculizumab, Ravulizumab, Zilucoplan, Tesidolumab, and SKY59).
2.2. Role of complement in MG (** )
The anti-AChR IgG antibodies bind to the AChRs at the neuromuscular junction, causing internalization of the receptors and activate complement which, by in situ MAC formation, leads to damage of the post-synaptic membrane and reduction of the AChR channels [Citation1,Citation7,Citation21,Citation22]. The role of complement in MG is supported by histopathological findings of IgG, C3, and C9 deposits at the neuromuscular junctions of MG patients; increased complement consumption during MG exacerbations, based on changes in the levels of various complement proteins in the patients’ sera [Citation24]; and increased capacity of the patients’ sera to deposit C3 fragments into sensitized targets, as measured by a quantitative C3 in vitro uptake [Citation25]. Animal models of experimental MG (EAMG), either by active immunization or passive transfer, have convincingly shown that complement deposition at the NMJ results in postsynaptic endplate destruction similar to the human disease [Citation26]. Animals deficient in C3 and C4 or with depletion of C5 or C6, have significantly lower incidence of developing EAMG or less severe disease because their IgG deposits at the Neuromuscular Junction (NMJ) do not lead to MAC formation [Citation27,Citation28]. Further, complement-inhibiting agents against C1q and C6, ameliorate the clinical signs of EAMG in active or passive transfer models by inhibiting MAC formation [Citation29,Citation30]. In contrast to AChR antibodies which are of IgG1-3 subclass, the anti-Musk antibodies which are of IgG4 subclass do not bind complement and do not cause end-plate destruction; instead, they are pathogenic causing dysfunction of the neuromuscular junction by interfering with AChR clustering through inhibition of signaling between agrin, lipoprotein receptor 4 (Lrp4) and Musk by blocking protein-protein interaction but not by antigen crosslinking, internalization and complement-mediated end-plate destruction as the IgG1-AChR antibodies do [Citation10–12]. Accordingly, Musk-MG patients do not respond to treatments that interfere with complement assembly, such as IVIg or anti-C3-C5 complement biologics, but robustly respond to agents that target B cells, such as rituximab [Citation1,Citation7,Citation31,Citation32].
2.3. Targets of anti-complement therapeutics
As highlighted in with asterisks, effective anti-complement agents will be those that inhibit the formation and in situ deposition of MAC by targeting key specific steps in the complement pathway. These include monoclonal antibodies or agents that target: (a) C1 subcomponents (*), now in development like ANX005 and Sutimilimab; (b) C3 (**) like compstatin and IVIg; and (c) C5 (***) targeted by at least five monoclonal antibodies, three of which, Eculizumab, Ravulizumab and Zilucoplan, have been already tested in MG with outstanding success, and they are either currently on the market or in phase III trials. The efficacy of these agents in MG is as follows:
2.3.1. Anti-C5(*** )
Eculizumab, a monoclonal antibody against C5
Eculizumab is the first drug in this category, approved for g MG in US but only for refractory g MG in Europe, that comprises up to 15% of all MG patients, based on a Phase-III placebo-controlled randomized trial (REGAIN) [Citation33]. Among the 125 AChR-positive participants, the eculizumab-randomized patients received 600 mg intravenously weekly for 4 weeks followed by 1200 mg every 2 weeks with the study’s primary end-point a change in MG-ADL score from baseline to week 26. Although no significant difference for the primary endpoint was observed, the MG-Activities of Daily Living (MG-ADL) and Quantitative-MG (QMG) scores significantly improved from baseline to week 26 in the 62 patients randomized to eculizumab compared to the 63 patients who received placebo [Citation33]. The MG exacerbations were also less frequent occurring in 10% of the eculizumab group, compared to 24% in the placebo group; further only 10% of the eculizumab-randomized patients required rescue therapy compared to 19% of the placebo group. Based on these data and the overall tolerance and safety, the drug gained FDA-approval. The benefit from Eculizumab is noticeable within the first 4–8 weeks, so unnecessary costly administrations can be avoided after early treatment initiation.
The patients who completed the REGAIN participated in the open-label extension (OLE) study, where 117 refractory-MG patients received treatment with 1,200 mg eculizumab every 2 weeks for a mean 22.7 month period [Citation34]. A 75% reduction of exacerbation rate and 80% reduction of MG-related hospitalizations rate were also noted with long-term safety and sustained efficacy throughout a 3-year period; further, 56% of the patients achieved minimal manifestation status or pharmacological remission [Citation34]. The placebo-randomized patients in the REGAIN study experienced rapid improvements in all assessment scores when treated with eculizumab in the OLE. Of interest, a significant proportion of patients in the OLE stopped or decreased their other immunosuppressive therapies, with prednisone decreased from baseline to the last assessment by 60.8%, the azathioprine by 89.1% and the mycophenolate mofetil by 56.0% [Citation35]. Those patients receiving chronic IVIg before entering the REGAIN study also showed sustained eculizumab efficacy over an 18-month period [Citation36]. Post-marketing analysis in 34 Japanese patients not included in OLE confirmed the drug’s safety and efficacy similar to the REGAIN trial [Citation37].
b. Ravulizumab, a humanized monoclonal antibody against C5 with increased half-life
Like eculizumab, Ravulizumab (Ultomiris) also binds with high affinity to C5 preventing the generation of complement activation products C5a and C5b-9 [Citation23]. In contrast to eculizumab however, Ravulizumab has increased half-life causing a more sustained complement inhibition requiring less frequent dosing (once every 8 weeks, instead of once every 2 weeks as for eculizumab) [Citation38]. Ravulizumab has gained FDA approval for Paroxysmal Nocturnal Hemoglobinuria (PNH). Released data from a phase III FDA-approved study in 175 MG patients showed treatment efficacy with significant change in MG-ADL score from baseline to 26 weeks with meaningful and statistically significant improvements in QMG score (https://clinicaltrials.gov/ct2/show/NCT03920293). On this basis, the drug was just approved by the FDA (April 2022) for the treatment of g MG as the first long acting C5 inhibitor. Ultomiris, following a loading dose, is given intravenously every 8 weeks and is expected to benefit a broader range of patients offering another option to MG patients potentially even those with milder symptoms.
c. Zilucoplan, a subcutaneously administered synthetic, macrocyclic peptide that binds C5
Zilucoplan binds to C5 with sub-nanomolar affinity and intercepts the MAC formation by inhibiting the cleavage of C5 into C5a and C5b [Citation23]. 44 patients with generalized MG in a phase II trial, received subcutaneous injection of either 0.3 mg/kg zilucoplan, 0.1 mg/kg zilucoplan or placebo on a daily basis for 12 weeks [Citation39]. Those patients randomized to the higher zilucoplan dose exhibited a mean reduction of 6.0 points in the QMG score from baseline, compared to 3.2 in the placebo group, and a 3.4 points reduction in the MG-ADL score compared to 1.1 point on placebo. The rapid and sustained improvements over 12 weeks was interpreted to suggest that maximal complement inhibition is necessary for pronounced disease suppression; these results along with good safety profile and absence of serious adverse effects led to an ongoing Phase III trial (RAISE) using the 0.3 mg/kg SC daily dosing (ClinicalTrials.gov Identifier: NCT04115293). Recently released data (16 February 2022) showed that Zilucoplan, a self-administered compound, in the completed Phase III trial demonstrated clinically meaningful and statistically significant improvements in both the primary and the secondary end points at week 12 with good tolerance and no unexpected safety issues. Based on these results, the sponsoring company publicly announced plans to regulatory filings for zilucoplan in g MG in the US, European Union, and Japan.
d. Pozelimab, a fully-humanized IgG4 monoclonal antibody designed to block C5
Pozelimab is currently undergoing clinical trials in MG (ClinicalTrials.gov Identifier: NCT05070858). The agent is used either alone or in combination with Cemdisiran, a small interfering RNA, targeting C5 designed to reduce the level of C5 mRNA in the liver, thereby reducing levels of circulating C5 protein, effectively inhibiting terminal complement pathway activity preventing the formation and deposition of MAC (C5-b9). Pozelimab is used sequentially with IV single dose following by sequential ascending to subcutaneous doses.
e. Future anti-C5 agents.
These include the Tesidolumab, a fully humanized monoclonal antibody against C5, and the SKY59, a C5 monoclonal antibody with a long half-life [Citation40]. Tesidolumab, like eculizumab, inhibits terminal complement activation and is being currently tested for the treatment of PNH (https://clinicaltrials.gov/ct2/show/NCT02534909.). SKY59, engineered to also inhibit FcRn, can be effectively recycled and recirculated [Citation40], ensuring long-lasting effects on neutralizing C5 requiring infrequent subcutaneous injections. In a small clinical trial, low-volume subcutaneous SKY59 was effective in eculizumab-treated patients with PNH suggesting promising effects for patients not sufficiently responding to eculizumab or with early-wearing off effects due to its more sustained anti-C5 action.
2.3.2. Anti C3 (**
C3, by being at the convergence point of all the complement activation pathways, is an obviously reasonable target in inhibiting the subsequent subcomponents of the complement cascade. Two agents affect C3, the high-dose Intravenous Immunoglobulin (IVIg) and the Compstatins.
IVIg has been shown to effectively bind C1q preventing the pathogenic antibodies from triggering the complement cascade [Citation12,Citation41,Citation42]. The F(ab’)2 region of the IgG within the IVIg participates in the neutralization of the C3a and C5a complement components, leading to uptake inhibition of C3b and C4b to the cell surface which prevents MAC formation and complement-mediated tissue injury [Citation24,Citation42]. IVIg is proven highly effective in a controlled trial of patients with Dermatomyositis, a complement-mediated microangiopathy, by intercepting the deposition of MAC on the endomysial capillaries enhancing neovascularization and muscle fiber regeneration [Citation43].
Compstatin is a generic name for various cyclic peptides that inhibit complement activation by binding to C3 and interfering with the function of convertase and C3 cleavage (). These family of peptides are currently in clinical trials in various diseases that involve dysregulated or excessive complement activation such as in age-related macular degeneration [Citation44] and PNH [Citation45].
2.3.3. Anti C1(*)
Two monoclonal antibodies, both of the IgG4 subclass, targeting the C1qC1r2C1s2 complex are currently tested in humans, the ANX005 against C1q and the Sutimlimab, against C1s.
a) ANX005, a humanized monoclonal antibody against C1q, functions as the initiating molecule of the classical complement cascade. A just completed phase 1b study evaluated the safety, tolerability and drug-drug interactions of ANX005 and IVIg in 14 patients with GBS who received a single dose of ANX005 75 mg/kg together with the standing IVIg treatment (ClinicalTrials.gov Identifier: NCT04035135), but the results are not yet reported. A phase 2/3 randomized, double-blind, placebo-controlled trial evaluating the safety and efficacy of ANX005 in 180 patients with GBS is ongoing (ClinicalTrials.gov Identifier: NCT04701164).
b)Sutimlimab (Enjaymo) In contrast to ANX005, sutimlimab specifically inhibits the classical pathway but does not inhibit the lectin and alternative pathways leaving intact the opsonic function of C1q. By blocking C1s, Enjaymo inhibits the activation of the complement cascade and the C1-activated hemolysis in adults with cold agglutinin disease (CAD) preventing hemolysis due to abnormal destruction of healthy red blood cells. Enjaymo has been now approved as the only treatment for adults with CAD due to its effectiveness in decreasing the need for red blood cell transfusions. In an in vitro study, sutimlimab also prevents complement-enhanced activation of autoimmune human B cells [Citation46] and could be of interest in future trials in B cell-mediated autoimmune neurological diseases, such as myasthenia gravis.
2.4. Safety of anti-complement therapeutics
Anti-C5 complement therapies now used for over than 10 years in paroxysmal nocturnal hemoglobinuria (PNH) and the least 3 years in Neurology have shown good safety profile even in patients concomitantly receiving immunosuppressants including anti-B cell agents like rituximab. Because formation of MAC is the primary defense against Gram-negative bacteria, especially Neisseria Meningitides, the main concern risk has been meningococcal infection for which vaccination is required prior to starting Eculizumab; this will be also applicable to other anti-complement agents when approved. A rare meningococcal infection related to serogroup B, which may not be protected by the meningococcal vaccination [Citation47], remains a remote risk for which some clinicians also consider antimicrobial prophylaxis. Because eculizumab does not accumulate in fetal blood or affect complement activation of the newborn and is not found in the breast milk, no safety concerns for its use in pregnancy have been raised. The drug has been very well tolerated with only higher incidence of mild headaches, nausea and urinary tract infections, especially in patients with significant comorbidities, but no incidence of meningitis. Eculizumab has been also safe in the setting of concomitant immunosuppressive use based on all the trials mentioned earlier [Citation33–38].
3. Modulation of the neonatal Fc receptor: a new category of antibody therapy (*** )
The neonatal Fc receptor (FcRn) plays a fundamental role in the IgG transport and recycling especially in reference to IgG catabolism in the lysosomes, extending the half-life and abundancy of IgG, as previously described [Citation1,Citation2,Citation7,Citation48,Citation49] (***). Agents that inhibit the function of FcRn cause rapid and sustained reduction of the circulating IgG including pathogenic antibodies in the lysosomes [Citation48,Citation49]. This family of immunotherapeutics has been especially advanced in MG with one drug in this group already FDA-approved, while two others are currently tested in phase-III trials:
Efgartigimod (ARGX-113), is a human IgG1 Fc fragment with increased binding affinity to FcRn that outcompetes endogenous IgG, preventing its recycling, and enhancing IgG degradation in the lysosomes. After a successful phase-II randomized trial in 24 MG patients that showed a rapid, within 2 weeks, long-lasting improvement in efficacy scales with reduction of AChR-antibody titers and good tolerance, a phase III (ADAPT) trial in 167 MG patients was conducted. Patients (most of them AChR-positive with a few seronegative) were randomized to 10 mg/kg efgartigimod in one-hour intravenous infusion, or placebo given in 4 weekly cycles followed by individualized 3 additional treatment cycles in 26 weeks. More efgartigimod-treated patients had clinically meaningful improvement in strength, MG-ADL and QMG scores associated with a mean reduction of IgG by 61.3% and of the AChR-Ab titers by 57.6% at week 4 [Citation50]. Importantly, the duration of response was more than 12 weeks in 34% and between 6 and 12 weeks in the majority of the rest. In the major phase III trial, Efgartigimod was very well tolerated with predominantly mild or moderate adverse events such as headaches, nasopharyngitis, and upper respiratory or urinary tract infection [Citation50], but without exerting signs of immunosuppression except of only a seemingly safe reduction of IgG level (hypogammaglobulinemia) but with a meaningful reduction of the AChR-antibody levels. This exciting novel therapeutic approach is now the first drug in this category to gain FDA-approval for MG [Citation51].
Rozanolixizumab, is a human anti-FcRn IgG4 monoclonal antibody with high affinity binding to FcRn; it is given subcutaneously resulting in reduced IgG levels by 75–90% from baseline. In a phase II study of 43 AChR and Musk-antibody-positive patients with generalized MG, a statistically significant improvement was noted in the MG-ADL associated with a 68% reduction in IgG and the AChR autoantibodies [Citation52]. These encouraging results led to an ongoing phase III study (NCT03971422). A recent news release revealed that the results from the phase III study showed effectiveness of Rozanolixizumab for g MG.
Nipocalimab, is a monoclonal antibody that binds to FcRn with picomolar affinity, promising high efficacy by ensuring occupancy of FcRn throughout the IgG recycling. A phase II study in MG showed that 51.9% of Nipocalimab-treated patients compared to 15% of the placebo, had a durable response based on MG-ADL scores at days 29 and 57 [Citation53], leading to a phase III trial (NCT03772587).
Batoclimab, is another anti-FcRn inhibitor also known as IMVT-1401; a phase 2 trial (NCT04346888) in 30 adults with moderate to severe g MG living in China showed the drug to lower the levels of disease-causing IgGs and eased MG symptoms going now to a phase III trial.
4. B cell-depleting agents in the therapy of MG (*)
Because B-cells play a fundamental role in antibody-mediated autoimmunity, including antibody production, antigen presentation, complement activation, and release of cytokines IL-1, IL-6, and IL-10, targeting B cells in MG patients can restore immune balance by suppressing antibody-mediated cytotoxicity [Citation1,Citation2,Citation54–56]. The family of anti-B cell therapies is rapidly expanding from only one drug, Rituximab, approved for systemic autoimmune diseases up to 2 years ago, to having now 3 new anti-B cell agents approved for autoimmune neurological disorders, such as Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders (NMOSD) [Citation55], all of which are also applicable to treating MG. Rituximab, a chimeric monoclonal antibody against CD20, depletes circulating B-cells, but not B-cells in the bone marrow and lymph nodes, or stem cells, pro-B cells and plasma cells [Citation54–56]. In many uncontrolled studies, Rituximab, has been effective in 50–70% of MG patients inducing sustained clinical improvement with excellent safety profile leading to tapering of other immunotherapies [Citation1,Citation3,Citation7,Citation57,Citation58]. Prospectively collected data from 72 patients conclusively showed that the time to remission was shorter for patients with new-onset disease, compared to conventional immunosuppressive therapies, when treated early with rituximab; these patients also showed fewer relapses with minimal, if any, need for additional immunotherapies suggesting that rituximab is more effective and safer than conventional immunosuppressants [Citation59]. Even in juvenile MG, retrospective 10-year data from a large, just published French series, showed effectiveness [Citation60]. In a series of 74 patients, 18 children with ocular and 56 children with g MG, rituximab used as a first-line immunosuppressant, showed better efficacy, tolerability, and safety with discontinuation of immunosuppressants and cortisone-sparing in 75% of juvenile myasthenia gravis patients [Citation60].
The success is impressive in Musk-MG [Citation61] which is an IgG4-mediated neurological disease unresponsive to IVIg [Citation11,Citation31,Citation61,Citation62]. A blinded, multicenter, prospective review showed that after a median follow-up of 3.5 years, 58% of patients treated with rituximab reached the primary outcome compared to 16% of controls requiring also less amount of prednisone [Citation61]. Marked reduction of IgG4-Musk antibodies is also seen after 2–7 months of Rituximab therapy [Citation62], coinciding with clinical remission and sustained improvement, suggesting that short-lived antibody-secreting cells are the main producers of Musk antibodies [Citation12,Citation62]. The reason why Rituximab is especially effective in Musk myasthenia is because Musk antibodies are of the IgG4 subclass which are non-inflammatory and non-complement fixing [Citation11,Citation12]. A phase II, but underpowered, placebo-controlled trial in 52 AChR-MG patients who received two cycles of rituximab 6 months apart, did not show statistically significant benefits [Citation63] because of the study design, that included patients with mild to moderate disease and based the primary response on the steroid dosage reduction [Citation63]. Rituximab was also shown to be effective in the few ocular MG patients [Citation32]. The drug is generally very well tolerated in all the aforementioned studies. We have not also seen any problems in more than 60 patients with various neurological autoimmunities we have treated the last 10 years; we always, however, check for immunoglobulin levels before the initial or the follow-up infusions to prevent further IgG reduction that may pose risks for infections. Also, no major safety data were reported in MS and NMOSD patients with four anti-B cell agents, as these data were carefully reviewed [Citation55]. Progressive multifocal leukoencephalopathy has been however reported in two patients who had also received long-term immunosuppressive therapies [Citation64].
Other anti-CD20/CD19 agents for future consideration in MG, extensively discussed recently [Citation55] include (a) Ocrelizumab, a humanized monoclonal antibody against CD20, approved for MS but not yet tried in MG; (b) Ofatumumab, that targets in addition to the large CD20 loop small epitopes closer to B-cell membrane causing more effective B-cell depletion [Citation55]. This benefit is exemplified by a patient with refractory MG who was unresponsive to IVIG, mycophenolate and rituximab, but normalized after two infusions with Ofatumumab exhibiting also a sustained depletion of circulating B-cells [Citation65]; (c) Obinutuzumab, which belongs to the third generation of anti-CD20′s and has been approved for chronic lymphocytic leukemia, causes profound depletion not only of peripheral B-cell lysis but also the lymphoid B-cells [Citation1,Citation54,Citation55]. A MG patient with CLL was almost cured after Obinutuzumab [Citation66]; (d) Ublituximab, another glycoengineered anti-CD20, currently in Phase-II MS trials; (e) Inebilizumab, an anti-CD19 monoclonal which also targets pro-B cells, plasmablasts and some plasma cells and has been now approved for Neuromyelitis Spectrum Disorders (NMOSD) [Citation55], may be a more attractive agent for a future trial in refractory MG, especially for Musk-MG . In fact, a trial with Inebilizumab is now ongoing in MG (NCT04524273); and f) Daratumumab, an anti CD38 agent that depletes plasma cells, as used in patients with multiple myeloma [Citation67]. CD38 is an integral membrane glycoprotein, present on early B and T cell lineages and activated B and T cells but not in mature resting peripheral lymphocytes [Citation67]. Because CD38 is also found on thymocytes, pre-B cells, germinal center B cells, and Ig-secreting plasma cells, it is an appropriate targeted therapy in MG by affecting antibody production. On this basis, a trial with Daratumumab is now ongoing in MG (NCT04159805) [Citation67].
5. How the new biologics is expected to change the treatment algorithm in MG
Although MG therapy remains patient-tailored according to age, personal or professional activities, tolerance and comorbidities, the new biologics do not seem at this point to substantially influence our therapeutic scheme for patients with mild or reasonably moderate AChR-MG. Crises and acute worsenings are still well controlled with plasmapheresis and IVIg and, after inducing a rapid but temporary improvement, the benefit is subsequently maintained with the conventional immunosuppressive therapies combined with low-dose corticosteroids, as outlined earlier, provided the patients do not have any further crises and the improvement is complete with good tolerance and quality of life.
This scenario does not however apply to patients with g MG who do not fully respond to the above therapies, exhibit poor satisfaction, intolerant side effects and several relapses, fulfilling overall the criteria of refractory g MG. For these patients, eculizumab or efgartrigimod are great options especially since both have now been FDA-approved. In counties however that these drugs are not widely available or still very expensive, rituximab is an excellent option, because of its accessibility, good tolerance and probably patients’ preference requiring infusions every 6–12 months or sometimes less often. For Musk-MG, rituximab is great a choice early in the disease. As the availability of eculizumab and efgartrigimod is however increasing and the cost becoming more accessible, the therapeutic algorithm is expected to change in their favor for the early stages of treatment because these drugs are clearly safer and preferable to immunosuppressants or high-dose steroids. When the subcutaneous or long-lasting forms of these biologics become also approved, their attractiveness and patients’ preference for earlier use is expected to substantially increase, changing the currently fluid algorithm toward their favor.
6. Expert opinion
New biologic therapies in MG are now changing the therapeutic treatment algorithm. On the personal level this is a monumental progress. When I started treating MG patients 40 years ago, we only had pyridostigmine, steroids and azathioprine with emerging, soon thereafter, plasmapheresis and IVIg for treating MG crises. New immunosuppressants, such as mycophenolate and Tacrolimus, became gradually available but their long-term safety profiles have been an issue especially for women of child-bearing age planning pregnancy; in older people comorbidities especially diabetes remains an issue for long-term steroid therapy. In the last 10 years, IVIg has been used by some practitioners for long-term therapy in spite of its transient benefits, prohibitive cost and unsubstantiated effectiveness in arresting disease progression with long-term use. Although we have done well saving lives, managing effectively the MG crises with plasmapheresis and IVIg, there has been a need for safe, effective, easy to use, accessible, and well tolerated long-term therapies. Further, none of the currently available immunotherapies have been approved specifically for MG or tested in large-scale prospective double blind studies as all agents are being used off-label. The scene is now getting brighter every year with targeted therapies that offer long-lasting benefits tested and confirmed, for the first time in MG therapeutics, in double-blinded, placebo controlled studies using validated scales. Most importantly, these agents are not ‘non-specific immunotherapies’ but they are targeted therapies in the form of monoclonal antibodies tailored to inhibit key pathogenic molecules responsible for the immunopathogenesis of the disease. They specifically target what is necessary to induce MG remission, namely, B cells, circulating autoantibodies, or complement, all fundamental players in antibody-mediated destruction of the AChR at the NMJ.
The biologics targeting complement are crucial because they inhibit the end plate disruption by not allowing the antibodies to fix MAC in situ, a monumental achievement based on the immunopathogenesis of MG offering now two FDA-approved drugs, one with short- 2 weeks- action (Soliris) and another (Ultomiris), with 8-weeks longer action. The biologics targeting FcRn are also major steps toward targeted immunotherapy because they safely lead to catabolism of IgG, including pathogenic antibodies, like we are doing plasmapheresis with an intravenous or even a subcutaneous injection. The results with both, anti-complement and FcRn, are impressive not only in suppressing pathogenic autoantibodies or make them less destructive, but also in achieving sustained remission with excellent tolerance. Not only two drugs, Eculizumab and Efgatrigimod, have been approved in the last 2 years, which is by itself monumental for MG, but there are close to 10 additional agents within these families of biologics that promise more effectiveness, tolerance and patients’ satisfaction. Trials with rituximab are also resulting in long-term remission especially in Musk-MG where the IgG4 antibodies do not fix complement and do not trigger inflammation but induce disruption of their antigenic target at the postsynaptic region by affecting protein-protein interaction [Citation10–12]. The third-generation of anti-B cells agents currently on the market, are additional tools toward targeted therapy promising sustained benefits, collectively fulfilling the author’s prior predictions that ‘the future of MG immunotherapies is not what used to be.’
The effects of these biologics do not only apply to MG but also to other neurological autoimmunities, like Aquaporin-positive Neuromyelitis Spectrum Disorders (NMOSD), where eculizumab and inebilizumab have also gained FDA-approval [Citation55,Citation68,Citation69]; anti-FcRn may also prove very promising because, as in MG, these antibodies are also pathogenic in NMOSD. Considering that complement plays a role in MOG-associated neuromyelitis (MOGAD) [Citation70], anti-complement therapies may also apply to this distinct but less common subset. I am personally excited that anti-complement therapies would have an impressive benefit in patients with dermatomyositis which is clearly a complement-mediated microangiopathy [Citation41,Citation43]. In contrast to recent efforts to divert the pathogenesis of dermatomyositis to alpha-interferon, a purely secondary and nonspecific downstream process as emphasized [Citation71], it is the inhibition of complement by IVIg in this disease that leads to impressive clinical improvement based on a controlled trial with restoration of muscle cytoarchitecture [Citation43]; the ongoing anti-complement trial in dermatomyositis (ClinicalTrials.gov NCT03669588) is therefore expected to be highly rewarding.
Witnessing such exciting benefits from the new therapies is a major satisfaction for clinical neurologists and neuroimmunologists with interest in immunotherapies, a field I have been personally serving for 40 years by conducting therapies with various immunomodulating agents and exploring the basis of underlying immunopathology. Most of all, it is a wonderful experience to see MG patients living normal lives without limitations with the belief that the future will be even better and the therapies easier to receive with less physical, emotional or economical burden.
Article highlights
C5 complement activation leads to in situ MAC formation which is fixed by the AChR antibodies at the neuromuscular junction, damaging the post-synaptic membrane.
Eculizumab by targeting C5 leads to sustained clinical remission of MG patients. On this basis, Eculizumab is the first ever FDA-approved drug in the chronic management of MG.
Ravulizumab (Ultomiris), an anti-C5 agent with longer-lasting effects compared to eculizumab, was now approved by the FDA for the treatment of generalized MG based on a phase III trial.
Zilucoplan is the first subcutaneous anti-C5 agent that seems also effective in MG with good tolerance and longer benefits, based on Phase II randomized trial.
Efgartigimod is the first biologic agent targeting FcRn that gained FDA-approved indication in treating MG based on its effectiveness and safety in a large controlled study.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
- Dalakas MC. Immunotherapy in myasthenia gravis in the era of biologics. Nat Rev Neurol. 2019;15:113–124.
- Schneider-Gold C, Hagenacker T, Melzer N, et al. Understanding the burden of refractory myasthenia gravis. Ther Adv Neurol Disord. 2019;12:1756286419832242.
- Dalakas MC. Progress in the therapy of myasthenia gravis: getting closer to effective targeted immunotherapies. Curr Opin Neurol. 2020;33(5):545–552.
- Evoli A. Myasthenia gravis: new developments in research and treatment. Curr Opin Neurol. 2017;30:464–470.
- Mantegazza R, and Antoni C. When myasthenia gravis is deemed refractory: clinical signposts and treatments strategies. Ther Adv Neurol Dis. 2018;11:1–11.
- Cortes-Vicente E, Alvarez-Velasco R, Segovia S, et al. Clinical and therapeutic features of myasthenia gravis in adults based on age at onset. Neurology. 2020;94:1171–1180.
- Evoli A, Spagni G, Monte G, et al. Heterogeneity in myasthenia gravis: considerations for disease management. Expert Rev Clin Immunol. 2021;17:761–771.
- McConville J, Farrugia ME, Beeson D, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55:580–584.
- Lauriola L, Ranelletti F, Maggiano N, et al. Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology. 2005;64:536–538.
- Koneczny I, Stevens JAA, De Rosa A, et al. IgG4 autoantibodies against muscle-specific kinase undergo fab-arm exchange in myasthenia gravis patients. J Autoimmunity. 2017; 77: 104–115.
- Huijbers MG, Zhang W, Klooster R, et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci U S A. 2013;110:20783.
- Dalakas MC. IgG4-mediated neurological autoimmunities: understanding pathogenicity of IgG4, ineffectiveness of IVIg and long-lasting benefits of anti-B cell therapies. Neurol Neuroimmunol Neuroinflammation. 2022;9:e1116.
- Wolfe GI, Kaminski HJ, Aban IB, et al. Long-term effect of thymectomy plus prednisone versus prednisone alone in patients with non-thymomatous myasthenia gravis: 2-year extension of the MGTX randomized trial. Lancet Neurol. 2019;18:259–268.
- Lee I, Kuo HC, Aban IB, et al. Minimal manifestation status and prednisone withdrawal in the MGTX trial. Neurology. 2020;95:e755–66.103.
- Gajdos P, Chevret S, Toyka KV. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev. 2012;12:CD002277.
- Dalakas MC. Update on intravenous immunoglobulin in neurology: modulating neuro-autoimmunity, evolving factors on efficacy and dosing and challenges on stopping chronic IVIg therapy. Neurotherapeutics. 2021November11;18:2397–2418
- Karelis G, Balasa R, De Bleecker JL, et al. A phase 3 multicenter, prospective, open-label efficacy and safety study of immune globulin (Human) 10% caprylate/chromatography purified in patients with myasthenia gravis exacerbations. Eur Neurol. 2019;81:223–230.
- Barth D, Nabavi Nouri M, Ng E, et al. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology. 2011;76:2017–2023.
- Beecher G, Anderson D, Siddiqi Z, et al. Subcutaneous immunoglobulin in myasthenia gravis exacerbation A prospective, open-label trial. Neurology. 2017;89:1–7.
- Dalakas MC. Future perspectives in target-specific immunotherapies of myasthenia gravis. Therap Adv Neurol Disorders. 2015;8(6):316–327
- Ruff RL, Lisak RP. Nature and action of antibodies in myasthenia gravis. Neurol Clin. 2018;36:275–291.
- Cetin H, Vincent A. Pathogenic mechanisms and clinical correlations in autoimmune myasthenic syndromes. Semin Neurol. 2018;38:344–354.
- Dalakas MC, Alexopoulos H, Spaeth P. Complement in autoimmune neurological disorders and emerging complement-targeted therapeutics. Nature Rev Neurol. 2020;16:601–617.
- Basta M, Illa I, Dalakas MC. Increased in vitro uptake of the complement C3b in the serum of patients with Guillain-Barré syndrome, myasthenia gravis and dermatomyositis. J Neuroimmunol. 1996;71:227–229.
- Howard JF Jr. Myasthenia gravis: the role of complement at the neuromuscular junction. Ann N Y Acad Sci. 2018;1412:113–128.
- Morgan BP, Chamberlain-Banoub J, Neal JW, et al. The membrane attack pathway of complement drives pathology in passively induced experimental autoimmune myasthenia gravis in mice. Clin Exp Immunol. 2006;146:294–302.
- Chamberlain-Banoub J, Neal JW, Mizuno M, et al. Complement membrane attack is required for endplate damage and clinical disease in passive experimental myasthenia gravis in Lewis rats. Clin Exp Immunol. 2006;146:278–286.
- Kusner LL, Sengupta M, Kaminski HJ. Acetylcholine receptor antibody-mediated animal models of myasthenia gravis and the role of complement. Ann N Y Acad Sci. 2018;1413:136–142.
- Souroujon MC, Brenner T, Fuchs S. Development of novel therapies for MG: studies in animal models. Autoimmunity. 2010;43:446–460.
- Farrugia ME, Vincent A. Autoimmune mediated neuromuscular junction defects. Curr Opin Neurol. 2010;23:489–495.
- Díaz-Manera J, Martínez-Hernández E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. 2012;78:189–193.
- Topakian R, Zimprich F, Iglseder S, et al. High efficacy of rituximab for myasthenia gravis: a comprehensive nationwide study in Austria. J Neurol. 2019;266:699–706.
- Howard JF, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalized myasthenia gravis (REGAIN): a phase 3, randomised, doubleblind, placebo-controlled, multicentre study. Lancet Neurol. 2017 Oct 20;16:976–986. Published Online.
- Muppidi S, Utsugisawa K, Benatar M, et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve. 2019;60:14–24.
- Nowak RJ, Muppidi S, Beydoun SR, et al. Concomitant Immunosuppressive therapy use in eculizumab-treated adults with generalized myasthenia gravis during the REGAIN open-label extension study. Front Neurol. 2020;11:556104.
- Jacob S, Murai H, Utsugisawa K, et al. Response to eculizumab in patients with myasthenia gravis recently treated with chronic IVIg: a subgroup analysis of REGAIN and its open-label extension study. Ther Adv Neurol Disord. 2020;13:1–7.
- Murai H, Suzuki S, Hasebe M, et al. Safety and effectiveness of eculizumab in Japanese patients with generalized myasthenia gravis: interim analysis of postmarketing surveillance. Ther Adv Neurol Disord. 2021;14:17562864211001995.
- Peffault de Latour R, Brodsky RA, Ortiz S, et al. Pharmacokinetic and pharmacodynamic effects of ravulizumab and eculizumab on complement component 5 in adults with paroxysmal nocturnal haemoglobinuria: results of two phase 3 randomised, multicentre studies. Br J Haematol. 2020;191:476–485.
- Howard JF, Nowak RJ, Wolfe GI, et al. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis results of a phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurol. 2020;77:582. Published online.
- Sampei Z, Haraya K, Tachibana T, et al. Antibody engineering to generate SKY59, a long-acting anti-C5 recycling antibody. PLoS One. 2018;13:e0209509.
- Basta M, Dalakas MC. High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J Clin Investig. 1994;94:1729–1735.
- Lunemann JD, Nimmerjahn F, Dalakas MC. Intravenous immunoglobulin in neurology–mode of action and clinical efficacy. Nat Rev Neurol. 2015;11:80–89.
- Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immunoglobulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993–2000.
- Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov. 2019;18:707–729.
- Ricklin D, Reis ES, Mastellos DC, et al. Complement component C3 - the “Swiss army knife” of innate immunity and host defense. Immunol Rev. 2016;274:33–58.
- Eskandary F, Jilma B, Mühlbacher J, et al. Anti-C1s monoclonal antibody BIVV009 in late antibody-mediated kidney allograft rejection-results from a first-in-patient phase 1 trial. Am J Transplant. 2018;18:916–926.
- Langereis JD, van den Broek B, Franssen S, et al. Eculizumab impairs Neisseria meningitidis serogroup B killing in whole blood despite 4CMenB vaccination of PNH patients. Blood Adv. 2020 Aug 11;4(15):3615–3620.
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. PubMed: 17703228.
- Dalakas MC, Spaeth PJ. The importance of FcRn in neuro-immunotherapies: from IgG catabolism, FCGRT gene polymorphisms, IVIg dosing and efficiency to specific FcRn inhibitors. Ther Adv Neurol Disord. 2021;14:1–7.
- Howard JF, Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled,phase 3 trial. Lancet Neurol. 2021;20:526–536.
- FDA News Release. FDA approves new treatment for Myasthenia Gravis. [cited 2021 Dec]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-myasthenia-gravis
- Bril V, Benatar M, Benatar M, et al. Efficacy and safety of rozanolixizumab in moderate-to-severe generalised myasthenia gravis: a phase 2 RCT. Neurology. 2020 Nov 20. https://doi.org/10.1212/WNL.0000000000011108. Epub ahead of print.
- Peter -H-H, Ochs HD, Cunningham-Rundles C, et al. Targeting FcRn for immunomodulation: benefits, risks, and practical considerations. J Allergy Clin Immunol. 2020;146:479–491.e5.
- Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nat Clin Pract Neurol. 2008;4(10):557–567.
- Stathopoulos P, Dalakas MC. Evolution of anti-B-cell therapeutics in autoimmune neurological diseases. Neurotherapeutics. 2022. https://doi.org/10.1007/s13311-022-01196-w
- Leandro MJ. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res Ther. 2013 Mar;15(1):1–8.
- Tandan R, Hehir MK, Waheed W, et al. Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve. 2017;56(2):185–196.
- Stieglbauer K, Pihler R, Topakian R. 10-year-outcomes after rituximab for myasthenia gravis: efficacy, safety, costs of in hospital care, and impact on childbearing potential. J Neurol Sci. 2017;375:241–244.
- Brauner S, Eriksson-Dufva A, Albert Hietala M, et al. Comparison between rituximab treatment for new-onset generalized Myasthenia Gravis and refractory generalized myasthenia gravis. JAMA Neurol. 2020 May 4;77:974.
- Molimard A, Gitiaux C, Barnerias C, et al. Rituximab therapy in the treatment of Juvenile Myasthenia Gravis: the French experience. Neurology. 2022. Publish Ahead of Print. doi:https://doi.org/10.1212/WNL.0000000000200288.
- Hehir MK, Hobson-Webb LD, Benatar M. Benatar M Rituximab as treatment for anti-MuSK myasthenia gravis. Neurology. 2017;89:1–9.
- Mariapaola Marino M, Basile U, Spagni G, et al. Long-lasting rituximab-induced reduction of specific—but not total—IgG4 in MuSK-positive Myasthenia Gravis. Front Immunol. 2020;11:613. doi: https://doi.org/10.3389/fimmu.2020.00613.
- Nowak RJ, Coffey CS, Goldstein JM, et al. NeuroNEXT NN103 BeatMG study team. Phase 2 trial of rituximab in acetylcholine receptor antibody-positive generalized Myasthenia Gravis: the BeatMG study. Neurology. 2021 Dec 2. doi:https://doi.org/10.1212/WNL.0000000000013121.
- Kanth KM, Solorzano GE, Goldman MD. PML in a patient with myasthenia gravis treated with multiple immunosuppressing agents. Neurol Clin Pract. 2016;6:e17–9.131.
- Waters MJ, Field D, Ravindran J. Refractory myasthenia gravis successfully treated with ofatumumab. Muscle Nerve. 2019;1–3. https://doi.org/10.1002/mus.26707
- Russell A, Yaraskavitch M, Fok D, et al. Obinutuzumab plus Clorambucil in a patient with severe myasthenia gravis and chronic lymphocytic leukemia J. Neuromuscul Dis. 2017;4. 251–257.
- Bonello F, Rocchi S, Baila G, et al. Safety of rapid daratumumab infusion: a retrospective, multicenter, real-life analysis on 134 patients with multiple myeloma. Front Oncol. 2022 Mar 14;12:851864.
- Pittock SJ, Berthele A, Fujihara K, et al., Eculizumab in aquaporin-4–positive neuromyelitis optica spectrum disorder. N Engl J Med. 381(7): 614–625. 2019.
- Wingerchuk DM, Fujihara K, Palace J, et al. Long‐term safety and efficacy of eculizumab in aquaporin‐4 IgG‐positive NMOSD. Ann Neurol. 2021;89(6):1088–1098.
- Keller CW, Lopez JA, Wendel E-M, et al. Complement activation is a prominent feature of MOGAD. Ann Neurol. 2021;90(6):976–982.
- Dalakas MC. Complement in autoimmune inflammatory myopathies, role of myositis-associated antibodies, COVID19-associations and muscle amyloid deposits. Expert Rev Clin Immunol. 2022 Mar 24;18: 413–423.