
ABSTRACT
Introduction
Generalized pustular psoriasis (GPP) is a rare, severe, clinically heterogeneous disease characterized by flares of widespread, noninfectious, macroscopically visible pustules that occur with or without systemic inflammation, and are associated with significant morbidity and mortality. Historically, GPP has been classified as a variant of psoriasis vulgaris (PV, or plaque psoriasis); however, accumulating evidence indicates that these are distinct conditions, requiring different treatment approaches.
Areas covered
In this perspective article we review evidence that supports the classification of GPP as distinct from PV.
Expert opinion
The histopathologic and clinical appearance of GPP is distinct from that of PV and fundamental differences exist between the two conditions in terms of genetic causes and expression-related mechanisms of disease development. GPP results from dysregulation of the innate immune system, with disruption of the interleukin (IL)-36 inflammatory pathway, induction of inflammatory keratinocyte responses, and recruitment of neutrophils. PV is driven by the adaptive immune system, with a key role played by IL-17. Considering GPP as a separate disease will enable greater focus on its specific pathogenesis and the needs of patients. Many treatments for PV have insufficient efficacy in GPP and a therapeutic approach developed specifically for GPP might lead to better patient outcomes.
Plain Language Summary
Generalized pustular psoriasis (GPP) is a rare disease. During episodes of worsening disease, the immune system attacks the skin. This causes large areas of skin to become red and painful, pus-filled blisters suddenly form. Some people with GPP have a history of another, more common, skin condition called psoriasis vulgaris (PV). People with PV develop patches of scaly, itchy skin. In the past, GPP was classed as a type of PV and treated with the same medicines. However, these medicines do not work well in GPP. Researchers now understand more about what causes GPP and how it differs from PV. GPP can cause medical problems throughout the body, leading to life-threatening complications. This means that people with GPP often need urgent medical treatment in hospitals. People with PV are mostly treated outside of hospitals. Any other medical problems are not usually due to PV itself. Researchers have found several genes that are altered in people with GPP and PV, and they differ between the two diseases. For example, changes in a gene called IL36RN are common in GPP but are not seen in PV. The skin of people with these two diseases also looks different under a microscope. Knowing more about GPP and how it differs from PV will help people with GPP to be diagnosed more quickly. It will also help researchers to develop new medicines specifically for GPP, so people can receive better treatment in the future.
Graphical Abtract

1. Introduction
Generalized pustular psoriasis (GPP) is a severe and potentially life-threatening condition [Citation1–3]. It is a rare disease, with annual prevalence estimates ranging from 1.76 per 1,000,000 persons in France in 2004 to 1.53 per 100,000 in Sweden in 2015, and point prevalence from 7.46 per 1,000,000 persons in Japan (1983–1989) to 9.1 per 100,000 persons in Sweden (2004–2015) [Citation4–6]. Although it can occur in children, GPP typically emerges during adulthood and is more common in women than men [Citation2,Citation7,Citation8]. GPP is a clinically heterogeneous systemic disease, characterized by flares involving widespread eruption of noninfectious or sterile, macroscopically visible pustules that can occur with or without systemic inflammation [Citation2,Citation3] and as either relapsing or persistent disease [Citation3,Citation9]. Flares are associated with significant morbidity and mortality, involving fever, sepsis, acute respiratory distress syndrome (ARDS), renal failure, and congestive heart failure [Citation2,Citation7,Citation10–12]; hospitalization is often required [Citation8,Citation13]. Patients with GPP frequently experience comorbidities, significantly reduced quality of life, and high medication use [Citation14–17]. Cohort studies have reported rates of mortality directly attributable to GPP, or its associated treatment, of 2–16% [Citation1,Citation4,Citation6,Citation7,Citation18–21].
Despite the severity of GPP, there are limited therapeutic options, and none have been specifically designed based on the disease pathogenesis. Treatment guidelines typically recommend cyclosporine, retinoids, infliximab, and methotrexate as first-line therapies, based on very weak evidence [Citation2,Citation22]. These treatments are often unsuitable for long-term use because they are associated with toxicities or are (or become) ineffective [Citation2,Citation22]. Biologic therapy has been reported to be effective in GPP, and several biologics, including certolizumab pegol, risankizumab, adalimumab, guselkumab, secukinumab, brodalumab, ixekizumab, and infliximab, have been approved for use in Japan, Taiwan, and Thailand. Although this is an important advance in GPP treatment options, current evidence is based on the results of small, single-arm trials using efficacy outcomes and time points derived from psoriasis vulgaris (PV) trials and not specifically designed for GPP [Citation2,Citation23–25]. No treatments have been approved specifically for the treatment of GPP flares and there is very weak evidence, if any, for the effectiveness of existing options for flare prevention [Citation2].
PV (also called plaque psoriasis) is a relatively common chronic disease, with an estimated prevalence that ranges from ~0.1% in East Asia to ~2% in Australia and Norway [Citation26]. It is the most common of the conditions that fall within the umbrella term psoriasis, accounting for around 90% of cases [Citation27,Citation28]. Skin symptoms include itching and pain, and as a chronic condition, PV has a significant and well-established impact on quality of life, often being associated with increases in anxiety and depression [Citation28,Citation29]. PV is also associated with psoriatic arthritis in 10–25% of cases, and other comorbidities, such as obesity, diabetes, cardiovascular disease, and inflammatory bowel disease (IBD), which further impair quality of life and increase morbidity and mortality, especially in its severe forms [Citation30,Citation31]. As PV is a common and well-characterized disease, multiple therapeutic options are available, from topical treatments for mild cases to biologics for severe disease [Citation32–35].
The term psoriasis, derived from the Greek term psora, meaning ‘itchy’ (and -iasis, meaning ‘condition’), has historically been used to describe a diverse variety of dermatologic conditions that are now considered etiologically distinct. Descriptions of psoriasis from as early as 200 CE have been re-categorized as leprosy and seborrheic dermatitis, as well as what would now be considered PV [Citation36]. Disease taxonomy will always be limited by the technology and knowledge of the time. This means that ongoing advances in science, medicine, and nosology must necessarily be paralleled by shifts and refinements in the classification of diseases. An excellent example of this can be seen with improvements in the treatment of breast cancer, which is now understood to be a group of heterogeneous diseases each with different clinical prognoses and therapeutic needs [Citation37]. Atopic dermatitis is another example of a heterogeneous condition (with some disease characteristics that overlap with PV) for which an increased understanding of molecular and inflammatory pathways is guiding the identification of therapeutic targets that can be used to develop individualized treatment strategies [Citation38,Citation39].
GPP was originally considered a variant or subtype of PV; however, accumulating evidence indicates that although 30–50% of patients with GPP may have a past history of PV [Citation5,Citation7,Citation18,Citation20,Citation40–43], the two diseases are distinct (). Even prior to the discovery of underlying genetic differences between the two conditions, researchers had proposed independent classifications for GPP or GPP subtypes, such as ‘generalized pustular dermatosis’ [Citation6]. More recently, a better understanding of the genetic markers and molecular pathways involved in the pathology of GPP and PV has led to a wider acceptance that these are likely to be separate entities [Citation13,Citation44–47]. Considering GPP as a disease in its own right, instead of as a severe form of PV, will enable greater focus on its specific pathogenesis and the needs of patients. A therapeutic approach developed specifically for GPP, rather than one based on the PV paradigm, might lead to better patient outcomes. Indeed, many treatments for PV have insufficient efficacy in GPP.
Table 1. Clinical, histological, and genetic differences between psoriasis vulgaris and generalized pustular psoriasis.
2. The classification and nomenclature of GPP
Until recently, the description of GPP as a rare subtype of PV was relatively common. Formal diagnostic tools, such as the International Classification of Diseases (ICD), position GPP as a subcategory of psoriasis; the ICD-10 coding for GPP, L40.1, falls within the broader psoriasis code but is distinct from PV (ICD-10 code L40.0) [Citation48]. However, in recent textbooks, classification of GPP has been refined as a member of a clinically heterogenous group of diseases collectively known as ‘pustular psoriasis,’ and a ‘distinctive acute variant’ within the spectrum of psoriatic diseases [Citation28,Citation49]. Published medical literature reporting cases of GPP also indicate that in a significant proportion of cases, patients with GPP do not have a past history of PV and thus it cannot be considered to be a consequence of PV [Citation1,Citation41–43]. This also implies that in individuals with both PV and GPP, the two conditions may be separate and require different treatment considerations.
GPP has various forms with differing presentation and severity [Citation2,Citation15,Citation28,Citation47]. The most typical form is acute (or von Zumbusch) GPP, associated with widespread pustules and systemic effects. Population-specific subtypes also present: GPP of pregnancy (previously known as impetigo herpetiformis) and neonatal/infantile/juvenile GPP. Furthermore, acute forms can be further divided into GPP with or without concomitant PV [Citation3,Citation47]. As such, it is incorrect to consider GPP part of a continuum of PV severity or an acute form, rather than a condition with its own subtypes and manifestations.
Along with GPP, other forms of pustular psoriasis include palmoplantar pustulosis (PPP) and acrodermatitis continua of Hallopeau (ACH). Based on its ‘clinical, epidemiological, genetic, and biological’ differences from PV, the International Psoriasis Council formally proposed in 2007 that PPP should not be included under the classification of PV [Citation50]. Based on evidence determined in the past 10 years, a similar recommendation may now be due for GPP. More recently, the European Rare and Severe Psoriasis Expert Network (ERASPEN) consensus statement delineated these pustular diseases from PV, noting that ‘primary pustules do not form part of the spectrum of PV except when pustules arise within or at the edge of psoriasis plaques’ and that ‘in these cases, the term to be used is “psoriasis cum pustulatione” (psoriasis with pustules) [and] this should not be considered pustular psoriasis’ [Citation3].
The ERASPEN consensus definition, which describes macroscopically visible sterile pustules on non-acral skin, notes that GPP pustules do not occur within psoriasis plaques and that GPP can occur with or without PV [Citation3]. The Japanese Dermatological Association (JDA) diagnostic definition of GPP, which requires the presence of systemic symptoms, extensive flush with multiple sterile pustules, neutrophilic subcorneal pustules, and repeated recurrence, excludes PV with transient pustules [Citation2]. JDA guidelines also indicate that concomitant PV may or may not be present [Citation2]. The separation of GPP from PV in these key guidelines recapitulates the importance of recognizing and treating GPP as an independent disease, linking accurate and specific diagnosis to treatment decisions and patient management recommendations.
Based on consensus opinions of global experts published between 2017 and 2019, GPP associated with known genetic mutations fulfills the criteria for an autoinflammatory disease (AID; a group of monogenic diseases primarily driven by inborn errors of the innate immune system and not deregulation of the adaptive immune responses), and is representative of an autoinflammatory keratinization disease (AIKD) [Citation46,Citation51–54]. This reflects the wider convergence to accurately classify GPP based on recent data, especially advances in our understanding of the genetic drivers of this disease. In line with this trend, the identification of mutations in the IL36RN gene (OMIM#614204) as a key driver of GPP in a high proportion of cases (described in more detail further on in this review) has prompted the use of the acronym DITRA (deficiency of interleukin 36 receptor antagonist) to describe these cases [Citation52,Citation55,Citation56]. Of note, neither PPP nor ACH contain the term psoriasis within their names, which reduces the potential for friction when reclassifying from a subtype of PV to a separate entity. The term GPP is likely to be too entrenched in the medical lexicon to be changed to a more accurate descriptor (e.g. as proposed by Ohkawara et al., to generalized pustular dermatosis [Citation6]); however, the inclusion of psoriasis in the name GPP may now be considered a misnomer, derived from historical classification and the initial description of presentation.
In April 2021, at a series of Delphi consensus meetings, a panel of patient advocacy group representatives and clinical experts concluded that GPP fits all the European Medicines Agency orphan disease criteria [Citation57]. Furthermore, the panel recommended that to emphasize the clinical and pathogenic distinction of GPP from other forms of psoriasis, future editions of the ICD should provide a better definition of GPP, and where it fits in the spectrum of psoriatic disease [Citation57].
3. GPP has a distinct histopathologic and clinical appearance
The appearance of the pustular lesions of GPP differs markedly from psoriatic plaques of PV (). PV is characterized clinically by the presence of discrete plaques with excess scale formation and is very rarely associated with visible pustules [Citation27,Citation58,Citation59]. The discrete nature of skin lesions leads to a vast difference in histologic characteristics compared with background (unaffected or non-lesional) skin () [Citation28,Citation60]. The PV lesion shows a markedly thickened epidermis (acanthosis), with deep, elongated rete ridges that constitute ‘psoriasiform hyperplasia’ () [Citation28,Citation60,Citation61]. There is a reduction in the granular layer with a consequence that maturation of keratinocytes is abnormal, and nuclei are retained in cells at the level of the stratum corneum [Citation28,Citation60,Citation61]. This cellular change, combined with a failure to secrete extracellular lipids, leads to weak cohesion of stratum corneum keratinocytes and more rapid shedding of surface cells. Thus, shedding of psoriasis ‘scales’ is a consequence of an abnormal differentiation program of keratinocytes in PV lesions. Microscopic neutrophilic infiltrates can be seen variably across different patients with PV and are not generally related to clinical disease severity (moderate or severe cases without neutrophilic infiltrates can be seen). Often, microscopic neutrophilic infiltrates are located in or just under the stratum corneum and are referred to as Munro’s microabcesses [Citation28,Citation61,Citation62].
Figure 1. Dermatologic presentation of PV (a) and GPP (b). PV is characterized by the presence of discrete scaly plaques, whereas the appearance of GPP is dominated by multiple coalescing white/yellowish pustules. Images courtesy of Dr. Burden. GPP, generalized pustular psoriasis; PV, psoriasis vulgaris.
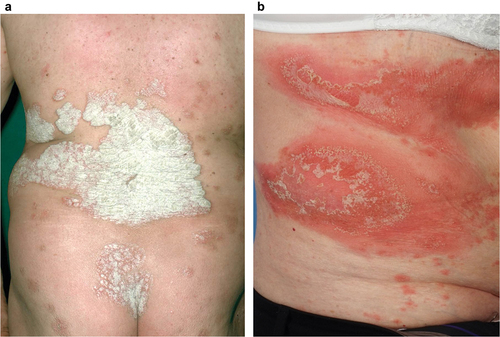
Figure 2. Histologic images of skin sections from patients with GPP (panels a to d) or PV (panels e to h) at comparable magnification, stained with hematoxylin and eosin (panels a, c, e, and g) and with anti-neutrophil elastase (panels b, d, f, and h). Panels a and b show edema of the epidermis with minimal acanthosis in a GPP lesion. Neutrophils are located at the surface in the subcorneal space (as would be seen in ‘lakes of pus’) and are abundant in the dermis. Panels c and d show moderate epidermal acanthosis in another case of GPP, with neutrophils located in mid-level epidermal vesicles, termed spongiform pustules of Kogoj, characteristic of a GPP diagnosis. The background, non-lesional skin from a patient with PV (panels e and f) has a normal-appearing epidermis and no neutrophils in the epidermis or dermis. A typical PV lesion (panels g and h) has marked epidermal acanthosis and rete pegs that are more elongated than in GPP. Neutrophil infiltration is variable in PV and may be largely absent. In this example, a few neutrophils can be seen in the mid-epidermis and stratum corneum, as shown by arrows. The epidermis in PV has much less edema than in GPP. Note the diffuse staining of neutrophil elastase in the epidermis of GPP (panels b and d), which indicates high levels of neutrophil degranulation with release of elastase into structural tissue of the epidermis. Bars in panels a, b, g, and h show 100 micrometers of length. Images courtesy of Dr. Krueger.GPP, generalized pustular psoriasis; PV, psoriasis vulgaris.
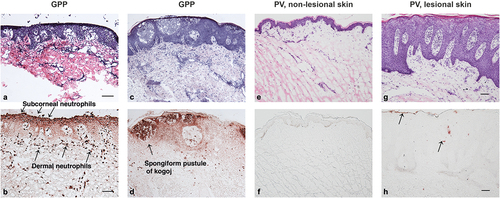
GPP has a clinical appearance that is dominated by the visible appearance of white/yellowish pustules that contain mainly neutrophils on microscopic examination (). In fact, the histologic appearance of GPP is dominated by the presence of neutrophils, which can appear as extensive accumulations in or just under the stratum corneum (with clinical correlate as ‘lakes of pus’) () [Citation2,Citation3,Citation63–65]. More discrete macroscopic pustules contain large vesicles packed with neutrophils that are seated in the mid-epidermis (spinous layer; ). This neutrophilic structure is much larger and more deeply invades into the epidermis than the Munro’s microabscess in PV. The deep-seating pustule in GPP has been named ‘the spongiform pustule of Kogoj’ and its presence is uniquely diagnostic of GPP [Citation2,Citation28]. Hence, pustulation, and not excess scaling of the epidermis, is one of the main differential features of GPP versus PV [Citation3].
4. GPP requires unique patient management considerations
4.1. Clinical manifestations and comorbidities
GPP is notable for its acute presentation with disease flares and considerable systemic involvement; the JDA criteria for a definitive diagnosis of GPP require the presence of systemic symptoms [Citation2], and both European and Japanese criteria highlight the relapsing nature of GPP [Citation2,Citation3]. A key clinical aspect of GPP is the potentially life-threatening nature of these flares, which require urgent, emergency intervention and frequent inpatient care [Citation7,Citation9,Citation21,Citation66]. It is estimated that a patient with GPP will require hospitalization for a flare at least once every 5 years [Citation7], with the duration of stay reported to be 10–14 days [Citation7,Citation9,Citation21]. A survey of dermatologists with experience of treating GPP in the USA indicated that 38% believed hospitalization for GPP was ‘somewhat common,’ with a further 21% stating it was ”very common” or ”always required” [Citation13]. Despite critical care, mortality rates directly attributable to GPP or its associated treatment are reported at between 2 and 16% [Citation1,Citation4,Citation6,Citation7,Citation18–21,Citation67]. In addition to acute symptoms, more than 80% of dermatologists surveyed indicated that patients with GPP experience residual disease between flares [Citation13].
Both cutaneous and extracutaneous manifestations of GPP contribute to its severe morbidity and potential mortality. During a GPP flare, patients present with systemic inflammation, not observed in PV, causing symptoms such as malaise, high-grade fever, and diarrhea [Citation68]. Inflammation is accompanied by laboratory abnormalities including raised C-reactive protein (CRP) and erythrocyte sedimentation rate, and leukocytosis with neutrophilia [Citation65,Citation68]. Notably, JDA guidelines highlight laboratory abnormalities (e.g. raised CRP, leukocytosis, elevated immunoglobulin, hypoproteinemia, and hypocalcemia) for use as diagnostic parameters and for the assessment of disease severity [Citation2]. Patients with GPP can experience direct extracutaneous neutrophilic inflammatory involvement, which is not seen in PV, resulting in conditions that include cholestasis, cholangitis, epigastric pain, arthritis, interstitial pneumonitis, oral lesions, and acute renal failure [Citation7,Citation19,Citation67,Citation69,Citation70]. Systemic neutrophilic GPP inflammation can rarely also lead directly to life-threatening conditions such as ARDS, cytokine storm, and congestive heart failure [Citation2,Citation4,Citation7,Citation12,Citation65,Citation68,Citation69]. Furthermore, microbial infections can occur within pustular skin [Citation69], with sepsis a potentially fatal complication [Citation4,Citation7,Citation65].
In contrast, PV is generally considered a chronic disease of the skin, typically managed in an outpatient setting [Citation28,Citation35]. Although patients with PV may also experience several systemic comorbidities, including immune-mediated inflammatory diseases, cardiovascular disease, and metabolic syndrome, in most cases etiological links between the conditions remain uncertain. It is likely that any association is multifactorial, involving side effects of treatments, and behavioral or lifestyle responses to living with PV in addition to shared genetic susceptibility and/or direct effects of PV [Citation28,Citation71]. In contrast to the life-threatening direct effects of GPP inflammation, deaths occurring in patients with severe PV tend to be related to these chronic comorbidities, such as cardiovascular disease [Citation72], rather than directly attributable to the psoriatic inflammation itself.
There is also a clear difference between GPP and PV in terms of presentation in pregnancy. Although complications have been reported during pregnancy in patients with PV, especially those with more severe disease, worsening of PV symptoms is possible but in a minority of cases [Citation73]. On the other hand, pustular disease is a distinct complication in pregnancy [Citation2,Citation7,Citation28,Citation40,Citation74]. Like acute GPP, GPP in pregnancy is potentially life-threatening to both mother and fetus in this particular situation [Citation2,Citation28,Citation75,Citation76]. Symptoms of GPP in pregnancy are similar to those of GPP flare outside of pregnancy, with possible systemic involvement in addition to complications relating to placental insufficiency [Citation7,Citation28,Citation76]. Worsening of GPP during pregnancy in patients with a preexisting GPP diagnosis has also been reported [Citation28,Citation77].
4.2. Systemic treatment considerations
As a relatively common disease, evidence and recommendations for the treatment of PV are well developed and provide multiple, approved therapeutic options for all disease grades [Citation31,Citation34,Citation35,Citation78]. The same is not true for GPP, in part because its rarity makes the conduct of clinical trials more challenging, but also because treatment is needed for both flare control and flare prevention [Citation2]. First-line therapy recommendations typically include cyclosporine, methotrexate, retinoids, and infliximab [Citation2,Citation22], although these are acknowledged to be based on insufficient evidence [Citation2].
Outside of Japan, Taiwan, and Thailand, no biologics are currently approved for use specifically in GPP. In Japan, the approval of biologics, including tumor necrosis factor (TNF)-α inhibitors, IL-17/IL-17 receptor (R) inhibitors, and IL-23 inhibitors, required trials in patients with GPP [Citation2,Citation25,Citation79–81], indicating that the authorities considered patients with GPP a distinct population and not part of a PV spectrum. The lack of approvals in other regions for biologics that are approved for use in PV supports this distinction.
Biologic therapies targeting cytokines such as TNF-α, IL-12, IL-23, and IL-17A have been reported to be effective in GPP [Citation2,Citation25,Citation79–82], and this is likely due to crosstalk between the cytokine pathways driving pathogenesis in GPP and PV. Although GPP likely originates in dysregulation of the IL-36 axis in epidermal keratinocytes with involvement of IL-17C (), this pathway can be activated by TNF or IL-17A (as potential triggers), but the level of activation for epidermal cytokines and chemokines in GPP is about 10-fold higher () than in PV [Citation83–86]. Thus, upstream inhibition of TNF or IL-17 (or IL-23, which regulates IL-17A) can likely attenuate the keratinocyte response to some extent, possibly enough to have visible improvement in GPP signs and symptoms [Citation83,Citation87]. However, the effectiveness of upstream inhibitors of IL-36 is uncertain in view of evidence derived from small (fewer than 12 patients), single-arm studies and case reports [Citation88,Citation89]. As such, interpretation of these data requires caution due to possible reporting bias, with only positive outcomes presented or published, and difficulties in assessing improvements from single-arm studies in a disease with self-remitting tendencies [Citation15,Citation59,Citation90,Citation91]. Furthermore, biologic therapies – notably TNF-α inhibitors – have been reported to induce pustular disease in patients being treated for other conditions, including PV [Citation92–96], highlighting potential disparities between beneficial (PV) and causative (GPP) mechanisms caused by these drugs. A single-center study indicated that drug survival for biologics was lower in patients with GPP than in those with PV [Citation97], although a separate report indicated that drug survival for patients with GPP was similar to that previously reported for PV [Citation42]. More data on larger patient cohorts are needed to accurately assess the effectiveness in GPP of biologics originally developed for the treatment of PV, and/or to confirm a differential effect of drugs on GPP and PV.
Figure 3. Fundamental variations in the Type 3 immune mechanism differentiate (a) PV and (b) GPP.
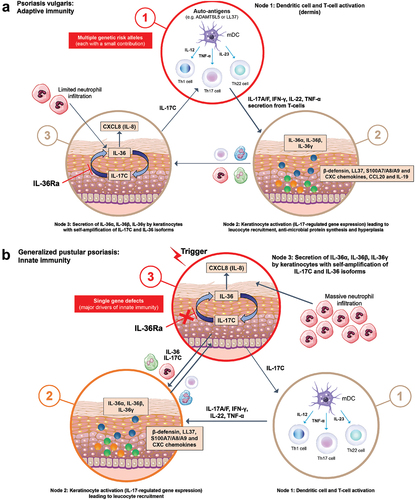
Table 2. Differentially expressed genes* relative to normal human skin in lesional skin from patients with PV and GPP [Citation84].
Based on evidence supporting a key role for the IL-36 pathway in GPP pathology, emerging therapies have targeted blockade of this pathway as a potential therapeutic approach specific to GPP. In the Effisayil™ 1 trial, efficacy and safety of the novel humanized anti-IL-36R monoclonal antibody, spesolimab, were evaluated in 53 patients with a GPP flare. One week after a single intravenous infusion, the proportion of patients with pustular clearance was significantly higher in the spesolimab arm (54%) than in the placebo arm (6%; p < 0.001), and this was sustained over the 12-week study [Citation98]. Similarly, early results with imsidolimab have shown a rapid and sustained reduction in area of erythema with pustules in 6 out of 8 patients with GPP [Citation99]. Further ongoing studies will add evidence on the long-term safety and effectiveness of this IL-36-targeted approach for both the treatment and prevention of GPP flares.
5. GPP and PV have different underlying genetic causes
The strongest evidence for a fundamental distinction between PV and GPP is provided by genetic analysis of patient populations, analysis of expression patterns between pustular and plaque disease, and by compelling mouse models of GPP/DITRA [Citation100]. These genetic analyses reveal fundamental differences in underlying genetic causes and expression-related mechanisms of disease development between GPP and PV.
5.1. Genetic drivers of GPP and PV are distinct
Multiple genetic abnormalities have been associated with the development of PV in a multigenic, complex model [Citation28,Citation101–103]. However, among these, the key allele is HLA*C0602, which encodes a major histocompatibility complex class I receptor involved in immune responses [Citation28,Citation101–103]. Mechanistic confirmation for the involvement of HLA*C0602 has been demonstrated through autoantigen presentation and subsequent triggering of PV [Citation103]. A meta-analysis of genome-wide association studies (GWAS) for PV published in 2017 identified a total of 63 psoriasis susceptibility loci in European-origin populations [Citation102]. Genes within these loci associated with causal biology are those involved in IL-23 and interferon (IFN) signaling pathways, T-helper (Th)17 cell differentiation and IL-17 responsiveness (e.g. IL23R, IL23A, IL12B, TYK2, TRAF3IP2), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-regulated signaling (e.g. NFKBIA, CARD14), and perturbation of the epidermal barrier (e.g. LCE3B/3C/3D) [Citation28,Citation102,Citation104–106]. Notably, some of the pathways affected by these genes have been used as targets for successful treatment of PV, for example with IL-23 and IL-17 inhibitors [Citation101].
In contrast to the polygenic abnormalities associated with the development of PV, and other frequent inflammatory diseases including psoriatic arthritis, the genetic architecture of familial cases of GPP appears to follow a monogenic, autosomal recessive Mendelian genetic model [Citation107]. In 2011, two pivotal reports using different methodologies described homozygous or compound double heterozygous loss-of-function (LOF) mutations in the IL36RN gene in most of the GPP cases they assessed [Citation44,Citation52], prompting one group to propose the acronym DITRA [Citation52]. Subsequent research identified further mutations in IL36RN and established defects in the IL-36 signaling pathway as a key driver of GPP pathogenesis [Citation56,Citation108,Citation109]. The IL-36 cytokine family, part of the IL-1 super-family, includes three agonists (activating ligands), IL-36α, IL-36β, and IL-36γ, and two antagonists, IL-36R antagonist (IL-36Ra) and IL-38 [Citation107,Citation110]. Binding of IL-36 activating ligands to IL-36R induces NF-κB and mitogen-activated protein kinase (MAPK), stimulating the release of pro-inflammatory cytokines that promote the activation of neutrophils, macrophages, dendritic cells, and T-cells [Citation52,Citation107]. IL36RN encodes IL-36Ra, which blocks the interaction of IL-36 ligands with IL-36R and prevents the exacerbation of inflammatory responses [Citation52,Citation107]. Thus, dysfunctional IL-36Ra leads to uncontrolled IL-36 pathway signaling and dysregulated production of inflammatory cytokines, such as IL-1, IL-6, and IL-8, which contributes to the development and maintenance of pustular disease via increased activation of keratinocytes [Citation44,Citation52,Citation107]. Altered IL36RN has been reported in ~21–24% of GPP cases overall [Citation41,Citation111]; however, for patients with GPP without PV, the proportion of cases with IL36RN mutations is considerably greater, at up to 82% [Citation56]. Notably, alterations in IL36RN are not associated with PV in patients without GPP [Citation112], and in patients with GPP, IL36RN mutations occur in the absence of mutations in key alleles associated with PV, including HLA*C0602 [Citation44]. The phenotypic consequences of IL36RN mutations are still being investigated. Patients with homozygous mutations, lacking expression of IL-36Ra (i.e. DITRA), present with high-grade fever during flares [Citation52]. Reports have also indicated earlier onset of GPP among patients with homozygous mutations compared with those carrying monoallelic mutations [Citation111]. Furthermore, some degree of correlation between the structural and functional impacts of IL36RN mutations and clinical severity has been demonstrated [Citation113]. These data provide a clear genetic basis for GPP independent of the presence of PV.
Additional genetic components of GPP have since been identified, providing evidence for the genetic basis of disease in patients without alterations in IL36RN. In Europe, approximately 11% of patients with GPP carry a mutation in AP1S3, which encodes a subunit of the adaptor protein 1 complex [Citation41], with some of these patients also having IL36RN mutations [Citation114]. Alterations in AP1S3 result in upregulated IL-1 signaling and overexpression of IL-36 cytokines (ligands), and inhibited keratinocyte autophagy [Citation114,Citation115], suggesting a likely role in GPP pathology. AP1S3 mutations have been reported in other pustular diseases, including PPP, ACH, and acute generalized exanthematous pustulosis (AGEP) [Citation116]. Studies have described mutations in CARD14 in small proportions of Chinese and Japanese patients with GPP, usually with concomitant PV [Citation117,Citation118]; however, a family with GPP showing autosomal dominant inheritance of CARD14 mutations in the absence of PV has also been reported [Citation119]. Given the scarcity of evidence currently available, the pathogenicity of CARD14 mutations has not yet been established for GPP, with murine models showing a PV-like rather than a GPP-like phenotype [Citation120,Citation121], whereas IL36RN transgenic mice have a GPP-like phenotype [Citation100]. The correlation between CARD14 mutations and the onset of GPP, in the absence of PV, therefore remains to be adequately defined. More recently, mutations in MPO (encoding myeloperoxidase) have also been identified as a driver of pustular disease, including GPP [Citation122,Citation123]. The mechanism for pathogenesis caused by myeloperoxidase deficiency has yet to be fully elucidated, although modifications in the activity of neutrophils and of proteases involved in the cleavage of IL-36 precursors to produce active IL-36 agonists have been reported [Citation122,Citation123]. In addition, an LOF heterozygous mutation in SERPINA3 (encoding a serine protease inhibitor) was identified in two patients with GPP and ‘significantly associated with GPP’ [Citation124]. As more genetic factors are identified, it becomes increasingly clear that the underlying cause of pustular disease, particularly GPP, is different from that of psoriatic plaques, and while there may be different genetic causes in individuals, it is striking that all pathways identified so far bisect at the level of IL-36.
5.2. Differences in gene expression in GPP and PV lesions
The identification of differing genetic drivers of GPP and PV is supported by several studies that show reproducible, distinct patterns of gene expression in skin biopsies from patients with these diseases.
Johnston et al. [Citation125] analyzed skin biopsy transcriptomes from patients with PV (n = 12) or GPP (n = 28, including 7 with concomitant PV) and healthy controls (n = 20). Notably, when transcriptomes of patients with both GPP and PV were assessed using unsupervised clustering analysis, they clustered with the GPP-only samples [Citation125]. Compared with healthy skin, GPP lesions and PV plaques showed a number of differentially expressed genes (DEGs), some that were unique to each disease (n = 295 in GPP, n = 670 in PV) and others that were common to both (n = 184). In addition, GPP expression profiles included more genes expressed by neutrophils and monocytes than PV expression profiles. Specifically, transcripts for IL-1β, IL-36α, and IL-36γ were more abundant in GPP lesions than in PV plaques, whereas transcripts for IL-17A, IL-22, IL-23p19, IFN γ, IL-18, and myxovirus resistance 1 were more abundant in PV plaques than in GPP lesions [Citation125].
Liang et al. [Citation126] assessed gene expression in skin biopsies from patients with GPP (n = 30), PPP (n = 17), and AGEP (n = 14) compared with healthy controls (n = 20). GPP lesions exhibited the largest number of DEGs (n = 2151 vs 461 for PPP and 197 for AGEP) and the most unique profile of DEGs (83.5% of DEGs being GPP-specific vs 42.3% for PPP and 0.5% for AGEP) [Citation126]. Protein ubiquitination demonstrated the greatest alteration in GPP; ubiquitination pathways are frequently dysregulated in inflammatory and autoimmune diseases [Citation126]. STEAP4 was a common DEG in all three diseases, and the investigators looked in more detail at the expression of the STEAP family of proteins in the pustular disease phenotypes compared with expression in PV plaques [Citation126]. Their finding that overexpression of STEAP1 and STEAP4 was seen across pustular psoriasis forms, including GPP, but not in PV, provided further robust evidence for the distinct nature of these diseases. The expression of STEAP1 and STEAP4 was clustered with that of inflammatory cytokines including IL-1, IL-36, and CXCL1/8, leading the authors to conclude that, ‘the distinct neutrophil-activating activities of STEAP1 and STEAP4 might underlie the pathologic alterations in patients with pustular psoriasis but not those with non-pustular psoriasis’ [Citation126].
Another transcriptome analysis compared molecular profiles of lesional and non-lesional skin from patients with GPP (n = 7) or PV (n = 17) with skin from healthy volunteers (n = 10) [Citation84]. Interestingly, non-lesional skin from patients with GPP showed 3683 DEGs, whereas non-lesional skin from patients with PV showed only 541 DEGs (), demonstrating considerable widespread non-focal skin involvement in GPP but not PV. There were also more DEGs in lesional skin from patients with GPP than in PV lesional skin (). In GPP lesional skin, 60% (n = 4115) of the DEGs did not overlap with DEGs found in PV lesional skin; among the 2759 DEGs that did overlap (40%), 1379 were upregulated in both diseases, but 789 (57%) of these showed higher dysregulation in GPP (p < 0.05). The largest differences were seen in genes involved in neutrophil-associated inflammation (CXCL1, CXCL8, CD177, CCL20) or connected with the Th1 axis (IL1B, IL36A) ().
Figure 4. Transcriptome analysis of skin biopsies from patients with PV and GPP compared with skin biopsies from healthy volunteers [Citation84]. The circles in the Venn diagrams illustrate the numbers of DEGs (genes with altered expression in the indicated biopsy compared with the healthy volunteer biopsy). Up-regulated genes are indicated by ↑, and down-regulated genes by ↓. The numbers of DEGs identified in both GPP and PV are illustrated by the overlap between the circles. (a) Biopsies of lesional skin from patients with PV or GPP compared with skin biopsies from healthy volunteers. Although a core of common DEGs is present in both GPP and PV lesions, most genes with altered expression in GPP are not altered in PV. (b) Biopsies of non-lesional skin from patients with GPP or PV, compared with skin biopsies from healthy volunteers, highlighting considerable dysregulation in non-pustular skin in patients with GPP.DEG, differentially expressed genes; GPP, generalized pustular psoriasis; HV, healthy volunteer; PV, psoriasis vulgaris.
![Figure 4. Transcriptome analysis of skin biopsies from patients with PV and GPP compared with skin biopsies from healthy volunteers [Citation84]. The circles in the Venn diagrams illustrate the numbers of DEGs (genes with altered expression in the indicated biopsy compared with the healthy volunteer biopsy). Up-regulated genes are indicated by ↑, and down-regulated genes by ↓. The numbers of DEGs identified in both GPP and PV are illustrated by the overlap between the circles. (a) Biopsies of lesional skin from patients with PV or GPP compared with skin biopsies from healthy volunteers. Although a core of common DEGs is present in both GPP and PV lesions, most genes with altered expression in GPP are not altered in PV. (b) Biopsies of non-lesional skin from patients with GPP or PV, compared with skin biopsies from healthy volunteers, highlighting considerable dysregulation in non-pustular skin in patients with GPP.DEG, differentially expressed genes; GPP, generalized pustular psoriasis; HV, healthy volunteer; PV, psoriasis vulgaris.](/cms/asset/ac4880c9-892a-4c4b-8cda-4d43593d31a8/ierm_a_2116003_f0004_oc.jpg)
Further evidence of the role of the IL-36 pathway in GPP was provided by Baum et al. [Citation127] who investigated the molecular changes in the skin and blood of patients with GPP flares after treatment with the novel humanized anti-IL-36R monoclonal antibody, spesolimab. Blocking IL-36R resulted in rapid normalization/downregulation of dysregulated genes associated with the IL-36 pathway, and this normalization was matched by clinical improvement [Citation127].
5.3. Pathogenic immune system pathways
The genomic and transcriptomic data discussed above correspond to functional differences observed in pathways driving the pathogenesis of GPP and PV (). The role of the adaptive immune system in driving PV is well established, leading PV to be described as ‘the best understood and most accessible human disease that is mediated by T-cells and dendritic cells’ [Citation128]. Lesional skin in PV contains Th1, Th17, and Th22 cells, activated by IL-23 mainly produced from myeloid dendritic cells in the skin [Citation128–130]. These Th cells produce several cytokines, such as IL-17A, IL-17F, IL-22, and IFNγ, resulting in hyperproliferation of keratinocytes, further inflammatory cytokine production, and subsequent chronic T-cell activation [Citation128–130]. The key role of the IL-17 pathway in the pathogenesis of PV is demonstrated by the high efficacy of therapies targeting IL-17A [Citation131].
In contrast to the adaptive immune system driving PV, GPP appears to be mainly the result of dysregulation of the innate immune system, fulfilling the definition of AID by experts in the field of this entity of mainly monogenic diseases, and also representative of the class of AIKDs [Citation46,Citation51]. In GPP, levels of Th17-related cytokines are significantly lower than in PV [Citation125], with IL-1 and IL-36 isoforms predominating. In the absence of functional IL-36Ra, as is common in GPP, there is massive amplification of activating IL-36 isoforms with induction of chemokines that recruit neutrophils, the dominant immune cell type in GPP [Citation130]. The resulting excessive neutrophil accumulation occurs through the activity of IL-36 pathways, potentially acting on keratinocytes to express the neutrophil chemoattractants CXCL1, CXCL2, and CXCL8 [Citation130,Citation132]. IL-36 signaling by keratinocytes in the granular layer induces expression of IL-17C, which creates a self-amplifying node as IL-17C also induces expression of IL-36 cytokines [Citation85,Citation86]. IL-17C can promote activation of IL-17A- or IL-17F-producing T-cells, and both these cytokines further stimulate synthesis of CXCL chemokines such as CXCL1, CXCL2, and CXCL8 [Citation133].
Despite these differing immune pathways underlying the pathogenesis of PV and GPP, crosstalk between the cytokine pathways mediating these inflammatory responses occurs. Therefore, cytokines such as TNF-α, IL-12, IL-23, and IL-17A, which are seen in PV, may not be key players in GPP pathogenesis, but do contribute to it [Citation125]; this results in, for example, the expansion of Th17 cells in GPP through the activity of the IL-36 pathway [Citation15]. Similarly, members of the IL-36 family are overexpressed in PV lesions in the absence of direct mutations in IL36RN, such as those seen in GPP [Citation132]. Combined with the genetic and expression data, this indicates the presence of independent pathways contributing to the development of either PV or GPP, albeit with necessary overlap in the cytokines and immune cells involved in disease-causing mechanisms.
6. Conclusion
GPP is a rare, orphan, AID that is characterized by acute, life-threatening flares and widespread systemic effects, and primarily driven by a deregulated innate immune system caused by single gene abnormalities. In contrast, PV is a highly prevalent, chronic inflammatory skin condition with typically non-pustular presentation primarily resulting from deregulated T-cell responses caused by the interaction of multiple gene abnormalities with environmental triggers. While the two conditions are related and may co-occur in patients, they do not represent a continuum of a single disease with GPP as an extreme form of PV. If GPP were to be classified today, with current knowledge and based on genetics, histopathology, clinical features, and therapeutic approaches, it would doubtlessly be considered a separate entity to PV. As understanding of the causes and unique nature of GPP has improved, the opportunity for appropriately targeted therapy to improve patient outcomes has increased.
7. Expert opinion
The IL-1 family of cytokines contains highly inflammatory molecules that can create severe inflammatory conditions if the balance of activation versus regulation is disturbed. The most common form of regulation is the production of cytokine-like receptor antagonists that bind to receptor subunits and prevent the binding of activating cytokine ligands. This mode of regulation may have developed as a consequence of redundancy in activating ligands for single receptors. For example, the IL-1R binds both IL-1α and IL-1β, with both cytokines inducing similar cellular effects, while IL-36R binds IL-36α, IL-36β, and IL-36γ, with similar activating effects. Signaling of IL-1R is restrained by IL-1Ra and signaling of IL-36R is restrained by IL-36Ra; receptor antagonists bind directly to the extra-cellular receptor to prevent receptor dimerization and signaling by the cognate activating ligands. A rare genetic mutation of the IL-1Ra prevents its function and the consequence is an inflammatory skin disease with pustulation and systemic inflammation termed Deficiency of IL-1 Receptor Antagonist (DIRA). Treatment of DIRA involves attenuation of IL-1α and IL-1β signaling with soluble receptor traps or other means. The disease most similar to DIRA is GPP, the subject of this review. There are clear genetic deficiencies in IL36RN, the gene that encodes IL-36Ra, in a sizable proportion of patients with GPP. GPP is a disease, like DIRA, that involves skin inflammation, pustulation, and systemic inflammation. In reference to DIRA, the acronym DITRA has been proposed for GPP related to inborn errors of the IL36RN gene. The latter, as well as other genetic defects identified in GPP, all converge in a deregulation of the innate immune system involving the IL-36 pathway as a key pathogenic circuit in GPP, making GPP a member of the so-called AID entity.
While IL-36 cytokines can activate many cell types that bear the IL-36R, there is a special relationship between skin as a barrier tissue and IL-36 cytokines. The epidermis, the outermost cutaneous tissue, protects from environmental microbial threats in part by the production of anti-microbial proteins (AMPs) in keratinocytes, a function that can be greatly accelerated by various inflammatory cytokines, and also by the ability of keratinocytes to synthesize chemokines that can recruit other immune cells into an inflammatory focus. Keratinocytes uniformly express IL-36R and can be activated in an autocrine or paracrine fashion by IL-36 cytokines synthesized by keratinocytes, with higher levels of cytokine production in the granular layer, which is relatively superficial and close to the external environment. IL-36 signaling in this location induces expression of the cytokine IL-17C, which creates a self-amplifying node as IL-17C also induces expression of IL-36 cytokines. IL-17C can promote activation of IL-17A- or IL-17F-producing T-cells and both these cytokines further stimulate synthesis of CXCL chemokines such as CXCL1 and CXCL8 (IL-8) that control neutrophil chemotaxis into the epidermis if microbes break through physical or anti-microbial protein barriers. In fact, pathogenic bacteria such as Staphylococcus aureus can directly induce IL-36 cytokine expression in human keratinocytes and this sets-off increased synthesis of the IL-36, IL-17C, CXCL chemokine response that would lead to neutrophil recruitment, producing another potent defense against microbial invasion. In the context of GPP, the IL-36/IL-17C node in keratinocytes and subsequent production of CXCL chemokines becomes aberrantly up-regulated, either through absence of IL-36Ra or by collective actions of other cytokines such as TNF-α, IL-1, IL-17A, IL-17C, or IL-17F that also stimulate synthesis of IL-36 cytokines in keratinocytes. The innate immune response, which encompasses keratinocyte production of AMPs and increases in neutrophils and other immune cells in the skin, becomes activated far beyond changes in PV, leading to massive recruitment of neutrophils that produce large, visible pustules with deep-seated structures that uniquely classify GPP or continuous sheets of neutrophils in a sub-corneal location (lakes of pus). If PV is a ‘trophic’ condition that increases epidermal mass and production of AMPs that protect against skin infection in a regulated fashion, GPP is in many ways opposite, as the physical skin barrier becomes disrupted by focal loss of stratum corneum and keratinocyte cohesion in viable layers of the epidermis is disrupted by massive edema as well as actions of neutrophil-derived proteases.
Although GPP has been classed as a subtype of PV because of historical reports, research has failed to find convincing arguments for GPP being part of the psoriatic disease spectrum in respect to genetics, transcription, clinical features, and response to treatment. There is now an increasing understanding among researchers – and in the literature – that the ‘psoriasis’ in the name GPP, in linking it to plaques, is a misleading misnomer. While there is some overlap in the epidemiology of GPP and PV, which is evident in a proportion of patients in whom a history of PV precedes GPP diagnosis, or in whom the two diseases co-exist, this may be related to intersection of the inflammatory networks driving disease pathogenesis. The divergent underlying genetics of the two diseases, however, strongly argues for their consideration as separate entities. The major arguments being that mutations in IL36RN, AP1S3, SERPINA3, and MPO have never been associated with PV, only with pustular disease, and that the dominant genetic model of familial cases of GPP is Mendelian and monogenic, while the dominant genetic model of PV is multigenic, complex, and non-Mendelian.
Article highlights
Historically, the description of generalized pustular psoriasis (GPP) as a rare subtype of psoriasis vulgaris (PV, also called plaque psoriasis) has been relatively common; however, a wealth of accumulating evidence indicates that GPP and PV are separate clinical conditions, requiring specific treatment approaches.
GPP and PV are distinct in terms of distribution on the body, and histopathologic and clinical appearance: PV is characterized by localized discrete plaques with excess scale resulting from abnormal differentiation of keratinocytes; GPP is characterized by widespread eruption of neutrophilic, non-infectious pustules.
GPP is notable for its acute presentation, with disease flares and complications resulting directly from neutrophilic inflammation, often requiring hospitalization; PV is a chronic disease of the skin with multifactorial comorbidities, typically managed in an outpatient setting.
Genetic drivers of GPP and PV also differ: many cases of GPP are familial and seem to follow a monogenic Mendelian model. GPP is frequently associated with mutations in IL36RN, which are not seen in PV. PV follows a complex polygenic model, with the key genetic driver being HLA*C0602, which is not associated with GPP.
Author contributions
All authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE) and made the decision to submit the manuscript for publication. The authors did not receive payment related to the development of the manuscript. All authors agree for the final version of the manuscript to be published.
Declaration of interest
H Bachelez declares paid consulting activities for AbbVie, Almirall, Anaptysbio, BIOCAD, Boehringer Ingelheim, Bristol Myers Squibb, Dermavant Sciences, Eli Lilly, Janssen, Kyowa Kirin, LEO Pharma, Novartis, and UCB; grant support from Boehringer Ingelheim, Bristol Myers Squibb, Janssen, LEO Pharma, Novartis, and Pfizer; and participation on a data safety monitoring board/advisory board for Avillion. J Barker declares having attended advisory boards and/or received consultancy fees and/or spoken at sponsored symposia, and/or received grant funding from AbbVie, Almirall, Amgen, AnaptysBio, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen, LEO Pharma, Novartis, Pfizer, Samsung, Sienna, Sun Pharmaceutical Industries, and UCB. A Burden declares paid consulting activities for AbbVie, Almirall, Boehringer Ingelheim, Celgene, Eli Lilly, Janssen, LEO Pharma, Novartis, and UCB. A Navarini declares being a consultant and advisor and/or receiving speaking fees and/or grants and/or serving as an investigator in clinical trials for AbbVie, Almirall, Amgen, Biomed, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Eli Lilly, Galderma, GlaxoSmithKline, LEO Pharma, Janssen-Cilag, MSD, Novartis, Pfizer, Pierre Fabre Pharma, Regeneron, Sandoz, Sanofi, and UCB. J Krueger declares consultancy/honoraria from AbbVie, Aclaris, Allergan, Almirall, Amgen, Arena, Aristea, Asana, Aurigene, Biogen Idec, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, Escalier, Galapagos, Janssen, MoonLake Immunotherapeutics, Nimbus, Novartis, Pfizer, Sanofi, Sienna Biopharmaceuticals, Sun Pharma, Target-Derm, UCB, Valeant, Ventyx; and grant support (to The Rockefeller University) from AbbVie, Akros, Allergan, Amgen, Avillion, Biogen, Botanix, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, Exicure, Innovaderm, Incyte, Janssen, Kyowa Kirin, Nimbus Lackshmi, Novan, Novartis, PAREXEL, Pfizer, Regeneron, UCB, and Vitae Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
One peer reviewer has participated as IP/SI and/or advisor and/or member of steering committee and/or invited speaker for IB, BMS, Abbvie, Novartis, Sandoz, Amgen, Janssen, and Lilly. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Acknowledgments
The authors would like to thank Sandra Garcet for her contribution to the transcriptome analysis of lesional and non-lesional skin from patients with GPP, PV and skin from healthy volunteers, which has been included in this review.
Additional information
Funding
References
- Ryan TJ, Baker H. The prognosis of generalized pustular psoriasis. Br J Dermatol. 1971;85(5):407–411.
- Fujita H, Terui T, Hayama K, et al., Japanese guidelines for the management and treatment of generalized pustular psoriasis: the new pathogenesis and treatment of GPP. J Dermatol. 2018;45(11): 1235–1270.
- Navarini AA, Burden AD, Capon F, et al., European consensus statement on phenotypes of pustular psoriasis. J Eur Acad Dermatol Venereol. 2017;31(11): 1792–1799.
- Augey F, Renaudier P, Nicolas J-F. Generalized pustular psoriasis (Zumbusch): a French epidemiological survey. Eur J Dermatol. 2006;16:669–673.
- Löfvendahl S, Norlin JM, Schmitt-Egenolf M. Prevalence and incidence of generalised pustular psoriasis in Sweden - a population-based register study. Br J Dermatol. 2022;186(6):970–976.
- Ohkawara A, Yasuda H, Kobayashi H, et al. Generalized pustular psoriasis in Japan: two distinct groups formed by differences in symptoms and genetic background. Acta Derm Venereol. 1996;76(1):68–71.
- Choon SE, Lai NM, Mohammad NA, et al. Clinical profile, morbidity, and outcome of adult-onset generalized pustular psoriasis: analysis of 102 cases seen in a tertiary hospital in Johor, Malaysia. Int J Dermatol. 2014;53(6):676–684.
- Morita A, Kotowsky N, Gao R, et al. Patient characteristics and burden of disease in Japanese patients with generalized pustular psoriasis: results from the Medical Data Vision claims database. J Dermatol. 2021;48(10):5–13.
- Kharawala S, Golembesky AK, Bohn RL, et al. The clinical, humanistic, and economic burden of generalized pustular psoriasis: a structured review. Expert Rev Clin Immunol. 2020;16:239–252.
- Abou-Samra T, Constantin JM, Amarger S, et al. Generalized pustular psoriasis complicated by acute respiratory distress syndrome. Br J Dermatol. 2004;150(2):353–356.
- Kluger N, Bessis D, Guillot B, et al. Acute respiratory distress syndrome complicating generalized pustular psoriasis (psoriasis-associated aseptic pneumonitis). J Am Acad Dermatol. 2011;64(6):1154–1158.
- Maehara Lde S, Mariano MM, Góis AF, et al. Acute respiratory distress syndrome as a complication of generalized pustular psoriasis. An Bras Dermatol. 2011;86(3):579–581.
- Strober B, Kotowsky N, Medeiros R, et al. Unmet medical needs in the treatment and management of generalized pustular psoriasis flares: evidence from a survey of Corrona Registry Dermatologists. Dermatol Ther (Heidelb). 2021;11(2):529–541.
- Sampogna F, Tabolli S, Söderfeldt B, et al. Measuring quality of life of patients with different clinical types of psoriasis using the SF-36. Br J Dermatol. 2006;154(5):844–849.
- Bachelez H. Pustular psoriasis: the dawn of a new era. Acta Derm Venereol. 2020;100(3):adv00034.
- Reisner DV, Johnsson FD, Kotowsky N, et al. Impact of generalized pustular psoriasis from the perspective of people living with the condition: results of an online survey. Am J Clin Dermatol. 2022;23(S1):1–7.
- Hanna ML, Singer D, Bender SD, et al. Characteristics of hospitalizations and emergency department visits due to generalized pustular psoriasis in the United States. Curr Med Res Opin. 2021;37(10):1697–1703.
- Choon SE, Navarini AA, Pinter A. Clinical course and characteristics of generalized pustular psoriasis. Am J Clin Dermatol. 2022;23(S1):21–29.
- Zelickson BD, Muller, SA. Generalized pustular psoriasis. A review of 63 cases. Arch Dermatol. 1991;127(9):1339–1345.
- Jin H, Cho HH, Kim WJ, et al. Clinical features and course of generalized pustular psoriasis in Korea. J Dermatol. 2015;42(7):674–678.
- Miyachi H, Konishi T, Kumazawa R, et al. Treatments and outcomes of generalized pustular psoriasis: a cohort of 1516 patients in a nationwide inpatient database in Japan. J Am Acad Dermatol. 2022;86(6):1266–1274.
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67(2):279–288.
- Thailand FDA. Lumicef® (brodalumab) product information 2019. updated 2022 Jul 22]. cited 2022 Jul 22. Available from: https://www.fda.moph.go.th/sites/oss/Drug%20Registration/Lumicef%20Subcutaneous%20Injection%20210%20mg%20Syringe_1C%2015051-61%20(NBC)/LUMICEF%20SUBCUTANEOUS%20_SPC(8-11-19).pdf
- Imafuku S, Honma M, Okubo Y, et al. Efficacy and safety of secukinumab in patients with generalized pustular psoriasis: a 52-week analysis from phase III open-label multicenter Japanese study. J Dermatol. 2016;43(9):1011–1017.
- Saeki H, Nakagawa H, Ishii T, et al. Efficacy and safety of open-label ixekizumab treatment in Japanese patients with moderate-to-severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis. J Eur Acad Dermatol Venereol. 2015;29(6):1148–1155.
- Parisi R, Iskandar IYK, Kontopantelis E, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ. 2020;369:m1590.
- Rendon A, Schäkel K. Psoriasis pathogenesis and treatment. Int J Mol Sci. 2019;20(6):1475.
- Burden A, Kirby B. Psoriasis and related disorders. Rook’s Textbook of Dermatology. 9 ed. New Jersey: John Wiley & Sons, Ltd; 2016.
- Martínez-Ortega JM, Nogueras P, Muñoz-Negro JE, et al. Quality of life, anxiety and depressive symptoms in patients with psoriasis: a case-control study. J Psychosom Res. 2019;124:109780.
- Alinaghi F, Calov M, Kristensen LE, et al. Prevalence of psoriatic arthritis in patients with psoriasis: a systematic review and meta-analysis of observational and clinical studies. J Am Acad Dermatol. 2019;80(1):251–65.e19.
- Nast A, Smith C, Spuls PI, et al. EuroGuiDerm guideline on the systemic treatment of psoriasis vulgaris - part 2: specific clinical and comorbid situations. J Eur Acad Dermatol Venereol. 2021;35(2):281–317.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80(4):1029–1072.
- Menter A, Gelfand JM, Connor C, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management of psoriasis with systemic nonbiologic therapies. J Am Acad Dermatol. 2020;82(6):1445–1486.
- Nast A, Smith C, Spuls PI, et al. EuroGuiDerm guideline on the systemic treatment of psoriasis vulgaris – part 1: treatment and monitoring recommendations. J Eur Acad Dermatol Venereol. 2020;34(11):2461–2498.
- National Insitute for Health and Care Excellence. Psoriasis: assessment and management. Clinical guideline. Published 2012 Oct 24, updated 2017 Sept 1. cited 2022 Jul 22. Available from: https://www.nice.org.uk/guidance/cg153
- Glickman FS. Lepra, psora, psoriasis. J Am Acad Dermatol. 1986;14(5):863–866.
- Ades F, Tryfonidis K, Zardavas D. The past and future of breast cancer treatment-from the papyrus to individualised treatment approaches. Ecancermedicalscience. 2017;11:746.
- Girolomoni G, de Bruin-Weller M, Aoki V, et al. Nomenclature and clinical phenotypes of atopic dermatitis. Ther Adv Chronic Dis. 2021;12:20406223211002979.
- Guttman-Yassky E, Krueger JG. Atopic dermatitis and psoriasis: two different immune diseases or one spectrum? Curr Opin Immunol. 2017;48:68–73.
- Ohata C, Tsuruta N, Yonekura K, et al. Clinical characteristics of Japanese pustular psoriasis: a multicenter observational study. J Dermatol. 2022;49(1):142–150.
- Twelves S, Mostafa A, Dand N, et al., Clinical and genetic differences between pustular psoriasis subtypes. J Allergy Clin Immunol. 2019;143(3): 1021–1026.
- Kromer C, Loewe E, Schaarschmidt M-L, et al. Drug survival in the treatment of generalized pustular psoriasis: a retrospective multicenter study. Dermatol Ther. 2021;34(2):e14814.
- Noe MH, Wan MT, Mostaghimi A, et al. Evaluation of a case series of patients with generalized pustular psoriasis in the United States. JAMA Dermatol. 2022;158(1):73–78.
- Onoufriadis A, Simpson MA, Pink AE, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet. 2011;89(3):432–437.
- Benjegerdes KE, Hyde K, Kivelevitch D, et al. Pustular psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Auckl). 2016;6:131–144.
- Akiyama M, Takeichi T, McGrath JA, et al. Autoinflammatory keratinization diseases. J Allergy Clin Immunol. 2017;140(6):1545–1547.
- Boehner A, Navarini AA, Eyerich K. Generalized pustular psoriasis - a model disease for specific targeted immunotherapy, systematic review. Exp Dermatol. 2018;27(10):1067–1077.
- World Health Organization. Chapter XII Diseases of the skin and subcutaneous tissue (L00-L99). World Health Organization; 2020. [cited 2022 Aug 31]. Available from: https://icd.who.int/browse10/2019/en#/XII
- Gudjonsson JE, Elder JT. Chapter 28. Psoriasis. Fitzpatrick’s Dermatology in General Medicine. 9 ed. New York: McGraw-Hill; 2019.
- Griffiths CEM, Christophers E, Barker JNWN, et al. A classification of psoriasis vulgaris according to phenotype. Br J Dermatol. 2007;156(2):258–262.
- Ben-Chetrit E, Gattorno M, Gul A, et al. Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): a Delphi study. Ann Rheum Dis. 2018;77(11):1558–1565.
- Marrakchi S, Guigue P, Renshaw BR, et al. Interleukin-36–receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365(7):620–628.
- Akiyama M. Early-onset generalized pustular psoriasis is representative of autoinflammatory keratinization diseases. J Allergy Clin Immunol. 2019;143(2):809–810.
- Navarini AA, Smith CH, Barker JN, et al. Reply. J Allergy Clin Immunol. 2019;143(2):810–811.
- Hospach T, Glowatzki F, Blankenburg F, et al. Scoping review of biological treatment of deficiency of interleukin-36 receptor antagonist (DITRA) in children and adolescents. Pediatr Rheumatol Online J. 2019;17(1):37.
- Sugiura K, Takemoto A, Yamaguchi M, et al. The majority of generalized pustular psoriasis without psoriasis vulgaris is caused by deficiency of interleukin-36 receptor antagonist. J Invest Dermatol. 2013;133(11):2514–2521.
- Koren J, Mburu S, Trigos D, et al. Generalised pustular psoriasis: the case for rare disease and orphan designation. Br J Dermatol. 2022; Online ahead of print. DOI:10.1111/bjd.21231.
- Oji V, Luger TA. The skin in psoriasis: assessment and challenges. Clin Exp Rheumatol. 2015;33(5 Suppl 93):S14–9.
- Uppala R, Tsoi LC, Harms PW, et al. “Autoinflammatory psoriasis”-genetics and biology of pustular psoriasis. Cell Mol Immunol. 2021;18(2):307–317.
- Boehncke W-H, Schön MP. Psoriasis. Lancet. 2015;386(9997):983–994.
- Raychaudhuri SK, Maverakis E, Raychaudhuri SP. Diagnosis and classification of psoriasis. Autoimmun Rev. 2014;13(4–5):490–495.
- Balan R, Grigoraş A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2019;6(3):59–68.
- Marrakchi S, Puig L. Pathophysiology of generalized pustular psoriasis. Am J Clin Dermatol. 2022;23(S1):13–19.
- Fujita H, Gooderham M, Romiti R. Diagnosis of generalized pustular psoriasis. Am J Clin Dermatol. 2022;23(S1):31–38.
- Ly K, Beck KM, Smith MP, et al. Diagnosis and screening of patients with generalized pustular psoriasis. Psoriasis (Auckl). 2019;9:37–42.
- Golembesky AK, Kotowsky N, Gao R, et al. PRO16 Healthcare Resource Utilization (HCRU) in patients with generalized pustular psoriasis (GPP) in Japan: a claims database study. Value Health Reg Issues. 2020;22:S98.
- Borges-Costa J, Silva R, Gonçalves L, et al. Clinical and laboratory features in acute generalized pustular psoriasis: a retrospective study of 34 patients. Am J Clin Dermatol. 2011;12(4):271–276.
- Varman KM, Namias N, Schulman CI, et al. Acute generalized pustular psoriasis, von Zumbusch type, treated in the burn unit. A review of clinical features and new therapeutics. Burns. 2014;40(4):e35–9.
- Hoegler KM, John AM, Handler MZ, et al. Generalized pustular psoriasis: a review and update on treatment. J Eur Acad Dermatol Venereol. 2018;32(10):1645–1651.
- Viguier M, Allez M, Zagdanski A-M, et al. High frequency of cholestasis in generalized pustular psoriasis: evidence for neutrophilic involvement of the biliary tract. Hepatology. 2004;40(2):452–458.
- Oliveira MDFSPD, Rocha BDO, Duarte GV. Psoriasis: classical and emerging comorbidities. An Bras Dermatol. 2015;90(1):9–20.
- Abuabara K, Azfar RS, Shin DB, et al. Cause-specific mortality in patients with severe psoriasis: a population-based cohort study in the U.K. Br J Dermatol. 2010;163(3):586–592.
- Vena GA, Cassano N, Bellia G, et al. Psoriasis in pregnancy: challenges and solutions. Psoriasis (Auckl). 2015;5:83–95.
- Tosukhowong T, Kiratikanon S, Wonglamsam P, et al. Epidemiology and clinical features of pustular psoriasis: a 15-year retrospective cohort. J Dermatol. 2021;48(12):1931–1935.
- Trivedi MK, Vaughn AR, Murase JE. Pustular psoriasis of pregnancy: current perspectives. Int J Womens Health. 2018;10:109–115.
- Namazi N, Dadkhahfar S. Impetigo herpetiformis: review of pathogenesis, complication, and treatment. Dermatol Res Pract. 2018;2018:5801280.
- Adachi A, Komine M, Hirano T, et al. Case of generalized pustular psoriasis exacerbated during pregnancy, successfully treated with infliximab. J Dermatol. 2016;43(12):1439–1440.
- Nast A, Gisondi P, Ormerod AD, et al. European S3-Guidelines on the systemic treatment of psoriasis vulgaris - Update 2015 - Short version - EDF in cooperation with EADV and IPC. J Eur Acad Dermatol Venereol. 2015;29(12):2277–2294.
- Yamasaki K, Nakagawa H, Kubo Y, et al. Efficacy and safety of brodalumab in patients with generalized pustular psoriasis and psoriatic erythroderma: results from a 52-week, open-label study. Br J Dermatol. 2017;176(3):741–751.
- Sano S, Kubo H, Morishima H, et al. Guselkumab, a human interleukin-23 monoclonal antibody in Japanese patients with generalized pustular psoriasis and erythrodermic psoriasis: efficacy and safety analyses of a 52-week, phase 3, multicenter, open-label study. J Dermatol. 2018;45(5):529–539.
- Wilsmann-Theis D, Schnell LM, Ralser-Isselstein V, et al. Successful treatment with interleukin-17A antagonists of generalized pustular psoriasis in patients without IL36RN mutations. J Dermatol. 2018;45(7):21–29.
- Arakawa A, Ruzicka T, Prinz JC. Therapeutic efficacy of interleukin 12/interleukin 23 blockade in generalized pustular psoriasis regardless of IL36RN mutation status. JAMA Dermatol. 2016;152(7):825–828.
- Furue K, Ito T, Tsuji G, et al. Psoriasis and the TNF/IL23/IL17 axis. Giornale Italiano di Dermatologia e Venereologia. 2019;154(4):418–424.
- Garcet S, Bachelez H, Baum P, et al. Distinct patterns of gene expression in skin biopsies differentiate generalized pustular psoriasis (GPP) from psoriasis vulgaris (PV). Society for Investigative Dermatology Anual Meeting, Portland, Oregon, May 18-21; 2022
- Chang SH, Reynolds JM, Pappu BP, et al. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. 2011;35(4):611–621.
- Friedrich M, Tillack C, Wollenberg A, et al. IL-36gamma sustains a proinflammatory self-amplifying loop with IL-17C in anti-TNF-induced psoriasiform skin lesions of patients with Crohn’s disease. Inflamm Bowel Dis. 2014;20(11):1891–1901.
- Muromoto R, Hirao T, Tawa K, et al. IL-17A plays a central role in the expression of psoriasis signature genes through the induction of IκB-ζ in keratinocytes. Int Immunol. 2016;28(9):443–452.
- Komine M, Morita A. Generalized pustular psoriasis: current management status and unmet medical needs in Japan. Expert Rev Clin Immunol. 2021;17(9):1015–1027.
- Wang WM, Jin HZ. Biologics in the treatment of pustular psoriasis. Expert Opin Drug Saf. 2020;19(8):969–980.
- Gooderham MJ, Van Voorhees AS, Lebwohl MG. An update on generalized pustular psoriasis. Expert Rev Clin Immunol. 2019;15(9):907–919.
- Takeichi T, Akiyama M. Generalized pustular psoriasis: clinical management and update on autoinflammatory aspects. Am J Clin Dermatol. 2020;21(2):227–236.
- Gregoriou S, Kazakos C, Christofidou E, et al. Pustular psoriasis development after initial ustekinumab administration in chronic plaque psoriasis. Eur J Dermatol. 2011;21(1):104–105.
- Kimura U, Kinoshita A, Haruna K, et al. Generalized pustular psoriasis-like eruptions induced after the first use of adalimumab in the treatment of psoriatic arthritis. J Dermatol. 2012;39(3):1931–1935.
- Wenk KS, Claros JM, Ehrlich A. Flare of pustular psoriasis after initiating ustekinumab therapy. J Dermatolog Treat. 2012;23(3):1–4.
- Caca-Biljanovska N, V’lckova-Laskoska M, Laskoski D. Successful management of ustekinumab-induced pustular psoriasis without therapy discontinuation. Acta Dermatovenerol Croat. 2013;21(3):202–204.
- Almutairi D, Sheasgreen C, Weizman A, et al. Generalized pustular psoriasis induced by infliximab in a patient with inflammatory bowel disease. J Cutan Med Surg. 2018;22(5):1463–1473.
- Kishimoto M, Komine M, Kamiya K, et al. Drug survival of biologic agents for psoriatic patients in a real-world setting in Japan. J Dermatol. 2020;47(1):33–40.
- Bachelez H, Choon SE, Marrakchi S, et al. Trial of spesolimab for generalized pustular psoriasis. N Engl J Med. 2021;385(26):2431–2440.
- Gudjonsson J, Reich A, Barker J, et al. Imsidolimab, an anti-IL-36 receptor monoclonal antibody, in the treatment of generalized pustular psoriasis: results from a phase 2 trial. European Academy of Dermatology and Venerology, Virtual; 2021. Sep-Oct 29-2.
- Blumberg H, Dinh H, Trueblood ES, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med. 2007;204(11):2603–2614.
- Capon F. The genetic basis of psoriasis. Int J Mol Sci. 2017;18(12):2526.
- Tsoi LC, Stuart PE, Tian C, et al., Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat Commun. 2017;8(1): 15382.
- Arakawa A, Siewert K, Stöhr J, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med. 2015;212(13):2203–2212.
- Strange A, Capon F, Spencer CC, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990.
- Stuart PE, Nair RP, Ellinghaus E, et al. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat Genet. 2010;42(11):1000–1004.
- Tsoi LC, Spain SL, Knight J, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44(12):1341–1348.
- Samotij D, Szczęch J, Reich A. Generalized pustular psoriasis: divergence of innate and adaptive immunity. Int J Mol Sci. 2021;22(16):9048.
- Setta-Kaffetzi N, Navarini AA, Patel VM, et al. Rare pathogenic variants in IL36RN underlie a spectrum of psoriasis-associated pustular phenotypes. J Invest Dermatol. 2013;133(5):1366–1369.
- Liu Z-J, Tian Y-T, Shi B-Y, et al. Association between mutation of interleukin 36 receptor antagonist and generalized pustular psoriasis: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore). 2020;99(45):e23068.
- Zhou L, Todorovic V, Kakavas S, et al. Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation. J Biol Chem. 2018;293(2):403–411.
- Hussain S, Berki DM, Choon S-E, et al. IL36RN mutations define a severe autoinflammatory phenotype of generalized pustular psoriasis. J Allergy Clin Immunol. 2015;135(4):1067–70.e9.
- Li M, Han J, Lu Z, et al. Prevalent and rare mutations in IL-36RN gene in Chinese patients with generalized pustular psoriasis and psoriasis vulgaris. J Invest Dermatol. 2013;133(11):2637–2639.
- Tauber M, Bal E, Pei XY, et al. IL36RN mutations affect protein expression and function: a basis for genotype-phenotype correlation in pustular diseases. J Invest Dermatol. 2016;136(9):1811–1819.
- Mahil SK, Twelves S, Farkas K, et al. AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J Invest Dermatol. 2016;136(11):2251–2259.
- Setta-Kaffetzi N, Simpson MA, Navarini AA, et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am J Hum Genet. 2014;94(5):790–797.
- Mössner R, Wilsmann-Theis D, Oji V, et al. The genetic basis for most patients with pustular skin disease remains elusive. Br J Dermatol. 2018;178(3):740–748.
- Zhou J, Luo Q, Cheng Y, et al. An update on genetic basis of generalized pustular psoriasis (review). Int J Mol Med. 2021;47(6):118.
- Sugiura K, Muto M, Akiyama M. CARD14 c.526G>C (p.Asp176His) is a significant risk factor for generalized pustular psoriasis with psoriasis vulgaris in the Japanese cohort. J Invest Dermatol. 2014;134(6):1755–1757.
- Takeichi T, Kobayashi A, Ogawa E, et al. Autosomal dominant familial generalized pustular psoriasis caused by a CARD14 mutation. Br J Dermatol. 2017;177(4):133–135.
- Mellett M, Meier B, Mohanan D, et al. CARD14 gain-of-function mutation alone is sufficient to drive IL-23/IL-17-mediated psoriasiform skin inflammation in vivo. J Invest Dermatol. 2018;138(9):2010–2023.
- Wang M, Zhang S, Zheng G, et al. Gain-of-function mutation of Card14 leads to spontaneous psoriasis-like skin inflammation through enhanced keratinocyte response to IL-17A. Immunity. 2018;49(1):66–79.e5.
- Vergnano M, Mockenhaupt M, Benzian-Olsson N, et al. Loss-of-function myeloperoxidase mutations are associated with increased neutrophil counts and pustular skin disease. Am J Hum Genet. 2020;107(3):539–543.
- Haskamp S, Bruns H, Hahn M, et al. Myeloperoxidase modulates inflammation in generalized pustular psoriasis and additional rare pustular skin diseases. Am J Hum Genet. 2020;107(3):527–538.
- Frey S, Sticht H, Wilsmann-Theis D, et al. Rare loss-of-function mutation in SERPINA3 in generalized pustular psoriasis. J Invest Dermatol. 2020;140(7):1451–5.e13.
- Johnston A, Xing X, Wolterink L, et al., IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol. 2017;140(1): 109–120.
- Liang Y, Xing X, Beamer MA, et al. Six-transmembrane epithelial antigens of the prostate comprise a novel inflammatory nexus in patients with pustular skin disorders. J Allergy Clin Immunol. 2017;139(4):1217–1227.
- Baum P, Visvanathan S, Garcet S, et al. Pustular psoriasis: molecular pathways and effects of spesolimab in generalized pustular psoriasis. J Allergy Clin Immunol. 2022;149(4):1402–1412.
- Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32(1):227–255.
- Ogawa E, Sato Y, Minagawa A, et al. Pathogenesis of psoriasis and development of treatment. J Dermatol. 2018;45(3):264–272.
- Schön MP. Adaptive and innate immunity in psoriasis and other inflammatory disorders. Front Immunol. 2019;10:1–14.
- Sawyer LM, Malottki K, Sabry-Grant C, et al. Assessing the relative efficacy of interleukin-17 and interleukin-23 targeted treatments for moderate-to-severe plaque psoriasis: a systematic review and network meta-analysis of PASI response. PLoS One. 2019;14(8):e0220868.
- Furue K, Yamamura K, Tsuji G, et al. Highlighting interleukin-36 signalling in plaque psoriasis and pustular psoriasis. Acta Derm Venereol. 2018;98(1):5–13.
- Furue M, Furue K, Tsuji G, et al. Interleukin-17A and keratinocytes in psoriasis. Int J Mol Sci. 2020;21(4):1275.