
ABSTRACT
Background
Tocilizumab, a recombinant monoclonal immunoglobulin G, targets the interleukin-6 receptor. MSB11456 is a proposed tocilizumab biosimilar.
Objectives
To assess pharmacokinetic equivalence of intravenous MSB11456 to US-licensed tocilizumab.
Research design and methods
In this double-blind, parallel-group, single-dose study, 128 healthy adults were randomized to a single one-hour 8 mg/kg IV infusion of either MSB11456 or US-licensed tocilizumab. Blood samples were collected pre-dose and at regular intervals up to day 48 post-dose. The primary endpoint pharmacokinetic parameter was analyzed using analysis of variance (ANOVA) model on the natural logarithm of the endpoint (AUC0–last), with treatment as a fixed effect. Immunogenicity and safety data were summarized descriptively.
Results
Subjects received either MSB11456 (N = 62) or US-licensed tocilizumab (N = 66). Pharmacokinetic bioequivalence, defined as 90% confidence intervals for the geometric least squares mean ratio entirely contained within the 80.00% to 125.00% equivalence limits, was demonstrated between MSB11456 and US-licensed tocilizumab for the primary and secondary pharmacokinetic endpoints. Anti-drug antibody responses, frequency of neutralizing antibodies against tocilizumab, and safety profiles showed no notable between-treatment differences. Safety was comparable between treatments.
Conclusions
Pharmacokinetic similarity of MSB11456 and US-licensed tocilizumab was demonstrated, with comparable immunogenicity and safety profiles, supporting MSB11455 as a biosimilar to US-licensed tocilizumab. The trial is registered at EudraCT, number 2019–003484-22.
Plain Language Summary
Tocilizumab is a biologic drug that is prescribed for autoimmune conditions such as rheumatoid arthritis in adults and arthritis in children where the cause is unknown. Because of the high cost of biologic drugs, alternate similar drugs are being designed and tested to ensure that they are as effective and as safe as drugs that are currently available. These new drugs are called biosimilars. MSB11456 is a proposed tocilizumab biosimilar. Our study tested how the pharmacokinetics, immunogenicity, and safety of intravenously administered MSB11456 compared to that of the already approved tocilizumab drug marketed in the US (US-licensed tocilizumab). One hundred and twenty-eight healthy adult volunteers received a one-hour 8 mg/kg intravenous infusion of either MSB11456 or US-licensed tocilizumab in this randomized, double-blind, parallel-group, single-dose study. Blood samples were taken before and at scheduled times during the study, up to 48 days after the first dose for analysis. In this study, we showed that the pharmacokinetics of MSB11456 were equivalent to the US-licensed tocilizumab. The safety and immune response to the drugs were also similar. These findings indicate that MSB11456 can be considered a biosimilar to tocilizumab. Biosimilars can reduce the cost of drugs by increasing competition and improve access to these, generally expensive, treatment options.
1. Introduction
Tocilizumab is a recombinant humanized monoclonal immunoglobulin G (IgG) antibody commonly used to treat moderate to severe rheumatoid arthritis (RA), systemic and polyarticular juvenile idiopathic arthritis, giant cell arteritis, T cell-induced severe or life-threatening cytokine release syndrome [Citation1], and more recently COVID-19 [Citation2]. Tocilizumab targets the interleukin-6 (IL-6) receptor; IL-6 is a pro-inflammatory cytokine responsible for the pathogenesis of many autoimmune diseases [Citation3] and a key driver in the inflammatory response [Citation4]. Tocilizumab dose-dependently binds with high affinity to the IL-6 receptor preventing binding by IL-6 [Citation3]. The pharmacokinetic (PK) profile is nonlinear and dose-dependent, and the elimination of tocilizumab is characterized by nonlinear and linear clearance pathways [Citation3,Citation5]. Tocilizumab is one of only a few approved biologic agents that inhibit the IL-6 pathway [Citation4,Citation6] and is approved by the US Food and Drug Administration (FDA) and the European Medicines Agency, as well as the regulatory agencies of other countries.
This randomized, double-blind, parallel-group study (APTURA II; EudraCT Number 2019–003484-22) was performed to determine whether a single one-hour 8 mg/kg intravenous (IV) infusion of MSB11456 (Fresenius Kabi SwissBioSim GmbH) and US-licensed tocilizumab (Actemra®) are bioequivalent in healthy adult subjects. Immunogenicity, safety, and tolerability of the two products were also compared.
2. Patients and methods
This was a randomized, double-blind, two-arm, parallel-group, single-dose study conducted at a single study center, Biokinetica S.A. Phase 1 Unit in Jozefow, in Poland, between 24 September 2020 and 29 January 2021. Subjects who met all eligibility criteria at screening and on day −1 were randomized in a 1:1 ratio prior to dosing on day 1, using interactive response technology, to receive a single one-hour 8 mg/kg IV infusion of either MSB11456 or US-licensed tocilizumab. The study was conducted during the COVID-19 pandemic. To protect subjects and to ensure the integrity of data, specific mitigation approaches were followed to minimize the risk of spreading severe acute respiratory syndrome 2 (SARS-CoV-2). All subjects provided written informed consent before study entry. The study was conducted in accordance with ethical principles of the International Council for Harmonization guideline for Good Clinical Practice and the Declaration of Helsinki, as well as with applicable local regulations (ICH/135/1995; EU directive 536/2014) and approved by an independent ethic committee.
2.1. Study population
One hundred and twenty-eight eligible subjects were included. These were healthy males or non-pregnant, non-breastfeeding females, aged ≥18 to ≤55 years with body mass index (BMI) ≥18.5 to ≤30.0 kg/m2, and in general good health based on a comprehensive medical assessment. Healthy was defined as an adequate hematological, renal, and hepatic function. Subjects were ineligible if they met any of the following exclusion criteria at screening or before randomization: history or presence of clinically significant atopic allergy or anaphylactic reactions; significant concurrent disease; confirmed or suspected active COVID-19 infection; history of tuberculosis, systemic fungal infection, serious infection (chronic or recurrent), alcohol or drug abuse, heavy smoking, malignancy, diverticulosis, or immunodeficiency; and previously exposed to tocilizumab or any other IL-6 acting drug (approved or investigational).
2.2. Assessments
The study included a screening period of up to 28 days and a 48-day treatment and follow-up period, which included admission to the study site on day −1, tocilizumab administration on day 1, discharge from the study site on day 3, and 12 ambulatory visits up to day 48. A total of 21 blood samples were collected from each subject for PK analyses: on day 1 before the start of infusion (0 hour) and 0.5, 1 (immediately after the end of infusion), 2, 4, 8, and 12 hours post-start of the infusion, and at 14 more scheduled times up until day 48 post-start of the infusion. PK serum concentrations were determined using a validated bioanalytical method.
Blood samples for immunogenicity, anti-drug antibodies (ADA) and neutralizing antibodies (NAb), were collected before the start of infusion on day 1, and on days 15, 29, and 48. Immunogenicity assessments used validated assays and were based on a multitiered approach: an electrochemiluminescence assay was used to detect ADA. All samples were screened and those putatively positive in the screening tier were tested in a confirmatory assay. All confirmed positive samples were further characterized by titration and then tested in a NAb cell-based assay to determine if the ADA against tocilizumab were neutralizing biological drug activity. Treatment-induced ADA status was defined as any post-dose sample being positive in the ADA confirmatory assay in any subject with an ADA-negative pre-dose sample, or a 1.808-fold increase (the minimum significant ratio) in titers from the pre-dose assessment to a post-dose assessment in any subject with an ADA-positive pre-dose sample.
Safety assessments included adverse event (AE) monitoring and severity grading. AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) version 23.0. The severity of all AEs was graded according to the National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE) Version 5.0. The CTCAE describes the severity of the Grades 1 through 5 based on the clinical guideline. Grade 1: mild, Grade 2: moderate, Grade 3: severe or medically significant, Grade 4: life-threatening, and Grade 5: death related to AE.
2.3. Study endpoints
The primary endpoint was the PK parameter area under the concentration–time curve (AUC) from time zero to the last quantifiable concentration (AUC0–last); maximum observed concentration (Cmax) and AUC from time zero to infinity (AUC0–inf) were included as secondary PK endpoints. AUC0–inf was not selected as a primary endpoint because the nonlinear PK characteristics of tocilizumab could impact the reliability of its estimation. Supplementary secondary endpoints included time to Cmax (tmax), terminal elimination half-life (t1/2), terminal elimination rate constant (λz), and clearance (CL).
Immunogenicity was measured as a secondary endpoint. Secondary safety endpoints, standardized according to regulatory requirements, included treatment-emergent adverse events (TEAEs), serious adverse events (SAEs), predefined adverse events of special interest (AESIs), which included hypersensitivity reactions events (NCI-CTCAE Grade ≥ 3) reported as serious and serious infections. Infusion site reactions (ISRs), vital signs, clinical laboratory values including hematology, chemistry, and urinalysis, and 12-lead electrocardiogram (ECG) were also considered.
2.4. Statistical analysis
A total of 128 healthy male and female subjects were to be randomized to ensure at least 116 evaluable subjects (58 evaluable subjects in each treatment group), assuming a maximum difference of 5% between MSB11456 and US-licensed tocilizumab, an inter-subject coefficient of variation (CV) not greater than 32%, and with an estimated dropout rate of approximately 10% to ensure at least 90% power to establish PK equivalence using conventional equivalence limits.
Two analysis sets were defined, i.e. the safety analysis set (all subjects who received any dose, partial or complete, of study treatment) for all safety analyses, and the pharmacokinetic analysis set (all subjects who received a complete dose of the study treatment, had sufficient concentration-time data to calculate the primary PK parameter, and who did not have any major protocol deviations or other events affecting PK assessment) for the analysis of PK parameters. PK parameters were calculated using standard noncompartmental methods with the validated software Phoenix® WinNonlin® Version 8.1 (Pharsight Corporation, a Certara Company, Princeton, New Jersey, USA). All other statistical analyses were validated and performed using SAS Version 9.4.
The primary parameter was analyzed with an analysis of variance (ANOVA) model on the natural logarithm of the endpoint (AUC0–last), with treatment as a fixed effect. Transformed back from the logarithmic scale, the geometric least-squares mean ratio (GMR) of the AUC0–last for MSB11456 to US-licensed tocilizumab, along with the corresponding 2-sided 90% confidence interval (CI) was estimated. PK equivalence between MSB11456 and US-licensed tocilizumab was declared if the 90% CI for the GMR for AUC0–last was contained entirely within the 80.00% to 125.00% equivalence limits.
Immunogenicity parameters included the number and percentage of subjects in the safety analysis set with ADAs and NAbs to tocilizumab during at least one post-baseline visit, presented by treatment group, overall, and visit. Summary statistics were also presented for median ADA titers by treatment group and visit.
Safety analysis parameters included the numbers and percentages of subjects with TEAEs, any SAEs, AESIs, local tolerability (including ISRs), clinical laboratory evaluations, and vital signs post-baseline. Subject baseline demographics and characteristics were summarized descriptively.
3. Results
Of 323 screened subjects, a total of 130 subjects were randomized. Of these, two subjects were withdrawn prior to dosing as an adverse event (AE) occurred after they were randomized. A total of 128 subjects received a single IV infusion of tocilizumab 8 mg/kg, of whom 62 received MSB11456 and 66 received US-licensed tocilizumab; all 128 subjects were included in the safety analysis set and PK analysis set and completed the study (). Participation in the study was not affected by any COVID-19-related study disruption, except for one subject who missed four visits and had to reschedule one visit due to contact with a person that tested positive for COVID-19.
Figure 1. Subject disposition.
Demographic characteristics were similar across both treatment groups. A total of 40 (31.3%) female and 88 (68.8%) male subjects between 18 and 54 years of age with a baseline BMI between 19.8 and 29.9 kg/m2 (baseline weight between 60.2 and 98.0 kg) participated in the study. All subjects identified as racially white, and none were of Hispanic or Latino ethnicity ().
Table 1. Subject demographics and characteristics at baseline by treatment groups. Safety analysis set.
3.1. Pharmacokinetics
3.1.1. Pharmacokinetic parameters of tocilizumab in serum
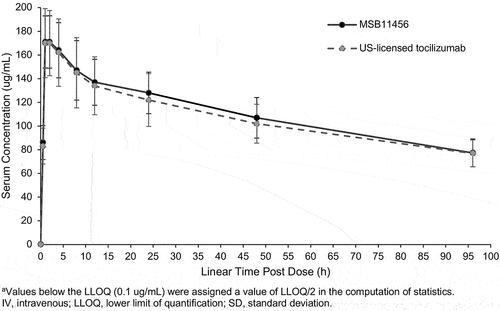
Following an 8 mg/kg IV infusion of MSB11456 or US-licensed tocilizumab, quantifiable drug concentrations were observed for all subjects at 0.5 hours post-dose. Tocilizumab concentrations increased quickly until they reached a peak concentration at approximately 2 hours post-dose, thereafter, gradually decreasing by a nonlinear, biphasic elimination (). Arithmetic mean serum concentration-time profiles of tocilizumab were almost superimposable following the infusion of MSB11456 and US-licensed tocilizumab.
Figure 2. Arithmetic mean (±SD) tocilizumab serum concentrationsa versus time on a linear scale following a one-hour 8 mg/kg IV infusion of MSB11456 and US-licensed tocilizumab in healthy subjects (pharmacokinetic analysis set).
Observed tocilizumab serum PK parameters are summarized by treatment in . The geometric least squares mean ratio (90% CI) of MSB11456 over US-licensed tocilizumab was 103.34% (98.53%, 108.37%) for the AUC0–last. The PK bioequivalence of MSB11456 versus US-licensed tocilizumab was demonstrated for the primary PK endpoint AUC0–last since the 90% CI of the geometric least squares mean ratio for AUC0–last of tocilizumab was entirely contained within the 80.00% to 125.00% equivalence limits. The 90% CIs for the geometric least squares mean ratio for the secondary PK endpoints Cmax and AUC0–inf were also contained within the 80.00% to 125.00% equivalence limits, supporting PK equivalence between MSB11456 and US-licensed tocilizumab (). Additional PK endpoints were all similar for MSB11456 and US-licensed tocilizumab ().
Table 2. Summary of tocilizumab serum pharmacokinetic parameters following a one-hour 8 mg/kg IV infusion of MSB11456 and US-licensed tocilizumab in healthy subjects (pharmacokinetic analysis set).
Table 3. Statistical analysis of the bioequivalence of a one-hour 8 mg/kg IV infusion of MSB11456 to US-licensed tocilizumab in healthy subjects (pharmacokinetic analysis set); primary and secondary pharmacokinetic parameters.
3.2. Immunogenicity
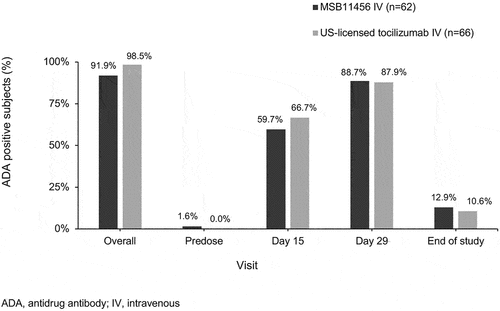
Only one subject, in the MSB11456 group, was ADA-positive prior to dosing. The overall post-infusion ADA positivity was similar between subjects who received MSB11456 (57 out of 62 [91.9%] subjects) and US-licensed tocilizumab (65 out of 66 [98.5%] subjects) (). For both MSB11456 and US-licensed tocilizumab, the ADA response was transient with a maximum proportion of ADA-positive subjects on day 29 (88.7% vs. 87.9%) and a smaller positive proportion at the end of the study (12.9% vs. 10.6%) as illustrated in . Median ADA titers were low, ranging from 60 to 240, with no noteworthy differences between the 2 treatment groups. The overall frequency of NAb against tocilizumab was low with no notable differences between MSB11456 (n = 4 NAb-positive subjects) and US-licensed tocilizumab (n = 8).
Figure 3. Bar chart for overall ADA and NAb positivity rate following a one-hour 8 mg/kg IV infusion of MSB11456 and US-licensed tocilizumab in healthy subjects (safety analysis set).
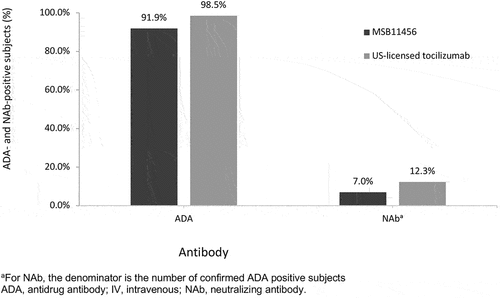
Figure 4. ADA incidence over time.
Within the ADA-positive population, PK parameters were similar between MSB11456 and US-licensed tocilizumab, which suggests that there was no difference in the impact of ADA on drug exposure for MSB11456 compared to US-licensed tocilizumab.
3.3. Safety
The safety profiles of a single IV 8 mg/kg infusion of MSB11456 and US-licensed tocilizumab were similar in healthy subjects. A total of 131 TEAEs were reported in 76 of 128 (59.4%) subjects; 61.3% of the MSB11456 group and 57.6% of the US-licensed tocilizumab group experienced at least one TEAE. Overall, in 41 out of 76 subjects (54%) with at least one TEAE, the most severe reported TEAE was Grade 1 or Grade 2 (). In both the MSB11456 and US-licensed tocilizumab group, the most commonly reported TEAEs were Grade 3 or 4 neutropenia (32.3% vs. 21.2%) and headache (16.1% vs. 13.6%); while all neutropenic events were considered treatment-related, headache events were considered unrelated. In the case of neutropenia and Grade 3 TEAE, the 95% CIsaround the observed proportion of subjects experiencing those events in the two treatment groups overlapped. Additionally, Grade 3 leukopenia was reported as 1.6% in the MSB11456 group and 4.5% in the US-licensed tocilizumab group. These events were transient and did not require treatment or intervention.
Figure 5. The most commona TEAEsb by preferred term and severity of TEAEs overall following a one-hour 8 mg/kg IV infusion of MSB11456 and US-licensed tocilizumab in healthy subjectsc (safety analysis set).
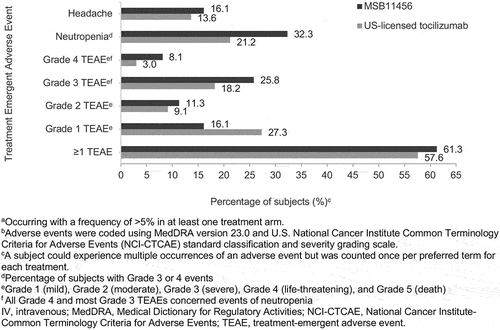
There were no distinguishing patterns in terms of nature, frequency, or resolution of TEAEs across treatments. No deaths, SAEs, AESIs, discontinuations, or infusion interruptions due to TEAEs were reported. There were eight ISRs (including infusion site bruising, rash, and swelling) in five subjects, with no notable differences between treatment groups (occurring in 4.8% of subjects who received MSB11456 vs. 3.0% who received US-licensed tocilizumab). All ISRs were Grade 1 (mild) in severity. Laboratory analyses (clinical chemistry, hematology, and urinalysis) revealed no discernible differences between treatment groups in terms of descriptive statistics or outliers. Likewise, there were no clinically significant abnormal values or differences noted in vital signs, electrocardiograms, or physician examinations between treatment groups.
4. Discussion
The primary objective of this study was to show PK equivalence of a single 8 mg/kg IV infusion of MSB11456 and US-licensed tocilizumab in healthy subjects was met. The 90% CI of the geometric least squares mean ratio for the primary PK endpoint AUC0–last, as well as for the secondary PK parameters Cmax and AUC0–inf were entirely contained within the predefined 80.00% to 125.00% equivalence limits. Other additional PK endpoints (tmax, t1/2, λz, and CL) were also similar between MSB11456 and US-licensed tocilizumab.
A parallel-group design was the preferred approach for this study considering the long terminal elimination half-life of tocilizumab following the IV route of administration and the potential influence of immunogenicity. The inclusion criteria were chosen to minimize variability not related to differences between the study drugs, including bodyweight limits (normal-overweight range [18.5–30.0 kg/m2]) as bodyweight can be one of the most influential factors of variability between subjects. The healthy subject model was regarded as a sufficiently sensitive and homogenous population, adequate to detect differences in PK and to allow extrapolation of the results to populations with indications for tocilizumab.
The PK of tocilizumab is influenced by many factors, including molecule size, binding location, absorption, distribution, excretion, and biotransformation characteristics [Citation7–9]. The nonlinear pattern of drug concentration and elimination of tocilizumab in this study is consistent with existing literature [Citation6,Citation10–12]. Concentration proportional increases in mean AUC and Cmax were expected and were consistent with those reported in other tocilizumab studies [Citation6,Citation11,Citation12] due to the desaturation of IL-6 receptor binding and rapid drug concentration decline. Mean t½ was similar between the two treatment groups (approximately 204 hours), which is comparable to that seen in other studies of IV tocilizumab 8 mg/kg (160 to 312 hours) [Citation6,Citation10,Citation12].
Two mechanisms may trigger immunogenicity with exposure to biotherapeutics, the recognition of non-self and lowered self-tolerance [Citation13]. In this study, immunogenicity was similar in terms of the number of subjects with positive ADA and/or NAb against tocilizumab, and ADA titers with both MSB11456 and US-licensed tocilizumab. Nearly all subjects had at least one positive ADA result post-dose (91.9% with MSB11456 vs. 98.5% with US-licensed tocilizumab). While median ADA titers were low, the incidence of ADA positivity was higher than in trials of tocilizumab IV monotherapy conducted in patients with rheumatoid arthritis [Citation14,Citation15]. The ADA assay used in this study was highly sensitive and drug tolerant, likely contributing to this higher incidence. Increased detection of immunogenicity has also been linked to patient-related and drug-development factors [Citation16–18]. In this study, the high immunogenicity incidence did not decrease tocilizumab efficacy [Citation14]. The FDA’s 2015 clinical review of biologics confirmed that 89% of all biologics studied have reported ADA positivity; however, ADA positivity affected PK in only 26% of the 121 approved biologic products [Citation19]. Of importance is that ADA positivity in this study was similar between treatment arms and was transient in most subjects. Furthermore, the frequency of NAb against tocilizumab was low (7% in subjects receiving MSB11456 and 12.3% in those receiving US-licensed tocilizumab), suggesting that the risk of secondary non-response with these drugs will be small.
The multifactorial nature of immunogenicity is well published, suggesting that there is not always a direct link between ADA and the clinical effects of the drug. A relationship between ADA status and PK outcomes could not be determined because of the low ADA incidence. The risk and consequence of developing ADA should be taken in the context of the safety, efficacy, and benefits of the treatment [Citation17,Citation20]. In this study, the presence of ADA did not result in symptoms of severe or serious hypersensitivity reactions, with no reported fever, rigors, or body aches noted. In addition, the absence of anaphylactic reactions indicated the absence of product-specific immunogenicity [Citation12,Citation13].
The safety profiles of MSB11456 and US-licensed tocilizumab, in terms of TEAEs, SAEs, AESIs, ISRs, were similar. The transient neutropenia noted in this study has also been reported in other studies [Citation12] and product guidelines [Citation21], with 7% to 15% of subjects receiving tocilizumab experiencing counts below 1 x 109/L. It should be noted that NCI-CTCAE v5 guidelines define neutropenia based on absolute neutrophil count values only, without accompanying clinical symptoms, and in this study, neither treatment nor intervention was required for those affected. A dose reduction to 4 mg/kg is recommended in certain dose-related neutropenia cases [Citation1].
Establishing the PK equivalence of MSB11456 with US-licensed tocilizumab, as demonstrated in this study, is of importance for the healthcare system. Recently, increased demand for tocilizumab in patients with COVID-19, coupled with supply and manufacturing issues, has led to a global shortage [Citation22]. The availability of biosimilars may help to alleviate this problem.
5. Conclusion
This randomized, single-center, double-blind, parallel-group study showed PK equivalence of MSB11456 with US-licensed tocilizumab after a single IV infusion of 8 mg/kg for one hour in healthy subjects. PK equivalence criteria were met for the primary and secondary PK endpoints. Safety profiles and immunogenicity were comparable between MSB11456 and US-licensed tocilizumab. This study therefore supports the biosimilarity of MSB11455 to US-licensed tocilizumab.
Declaration of interest
M Ullmann, C Petit- Frere, J Monnet, and A Illes are employees of Fresenius Kabi SwissBioSim GmbH. C Dagres is a former Fresenius Kabi SwissBioSim GmbH employee. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
Conception of the work: A. Illes.
Design of the work: M.Ullmann, C. Petit-Frere, A.Illes.
Acquisition of data for the work: M.Ullmann, M. Tomaszewska-Kiecana, C. Petit-Frere.
Analysis of data for the work: M.Ullmann, C. Petit-Frere, J. Monnet.
Interpretation of data for the work: M. Ullmann, C. Petit-Frere, J. Monnet, C. Dagres, A. Illes.
Drafting of the manuscript: C. Petit-Frere.
Critical revision of the manuscript for important intellectual content: M. Tomaszewska-Kiecana, M. Ullmann, C. Petit-Frere, J. Monnet, C. Dagres.
All authors have participated sufficiently in the work to agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors have given approval of the manuscript to be published.
Research involving human participants: ethics approval
Before initiating the study, the investigator obtained written and dated approval from the Independent Ethics Committee, Komisja Bioetyczna przy Okręgowej Izbie Lekarskiej w Warszawie, for the study protocol and its amendments, written informed consent form (ICF), any ICF updates, subject recruitment material, subject information sheets, and other subject-facing material.
This study was conducted in accordance with the Note for Guidance on Good Clinical Practice (GCP) International Council on Harmonisation (ICH) Harmonised Tripartite Guideline E6 (R2) from the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use ICH/135/1995, and requirements for the conduct of clinical studies as provided in the EU Directive 536/2014; the general guidelines indicated in the Declaration of Helsinki; and all applicable regulatory requirements.
Informed consent
Informed consent was obtained in compliance with the applicable regulatory requirements and adhered to GCP and to the ethical principles that have their origin in the Declaration of Helsinki. Each subject signed and dated two identical original ICFs, of which one was retained at the study site and the other was provided to the subject prior to participation. The trial is registered at EudraCT, number 2019-003484-22.
Acknowledgments
The sponsors acknowledge the participation of all healthy volunteers and the study site personnel involved in the clinical trial. Medical writing support was provided by Clare Koning and Caroline Spencer (Rx Communications, Mold, UK), funded by Fresenius Kabi SwissBioSim.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Genentech. Actemra®(tocilizumab) injection, for intravenous or subcutaneous use. Highlights of prescribing information. 2022. South San Francisco (CA). [ Online]. Online. [Cited 2022 Nov 2]. Available from: https://www.gene.com/download/pdf/actemra_prescribing.pdf
- World Health Organization (WHO). Prequalified Medicinal Products. Tocilizumab. Reference BT-CV001 (a). World Health Organization Prequalification Team (10 February 2022).
- Shetty A, Hanson R, Korsten P, et al. Tocilizumab in the treatment of rheumatoid arthritis and beyond. Drug Des Devel Ther. 2014;8:349–364.
- Crisafulli S, Isgrò V, La Corte L, et al. Potential Role of Anti-interleukin (IL)-6 Drugs in the Treatment of COVID-19: rationale, Clinical Evidence and Risks. BioDrugs. 2020;34(4):415–422.
- Navarro-Millán I, Singh JA, Curtis JR. Systematic review of tocilizumab for rheumatoid arthritis: a new biologic agent targeting the interleukin-6 receptor. Clin Ther. 2012;34(788–802.e3):788–802.e3.
- Song SN, Yoshizaki K. Tocilizumab for treating rheumatoid arthritis: an evaluation of pharmacokinetics/pharmacodynamics and clinical efficacy. Expert Opin Drug Metab Toxicol. 2015;11(2):307–316.
- Dua P, Hawkins E, Van Der Graaf P. A Tutorial on Target-Mediated Drug Disposition (TMDD) Models. CPT: Pharmacomet Syst Pharmacol. 2015;4:324–337.
- Smit C, De Hoogd S, Rjm B, et al. Obesity and drug pharmacology: a review of the influence of obesity on pharmacokinetic and pharmacodynamic parameters. Expert Opin Drug Metab Toxicol. 2018;14(3):275–285.
- Leung E, Crass RL, Jorgensen SCJ, et al. Pharmacokinetic/Pharmacodynamic Considerations of Alternate Dosing Strategies of Tocilizumab in COVID-19. Clin Pharmacokinet. 2022;61(2):155–165.
- Nishimoto N, Terao K, Mima T, et al. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti–IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. 2008;112(10):3959–3964.
- Fettner S, Mela C, Wildenhahn F, et al. Evidence of bioequivalence and positive patient user handling of a tocilizumab autoinjector. Expert Opin Drug Deliv. 2019;16(5):551–561.
- Zhang H, Li X, Liu J, et al. A randomized phase-I pharmacokinetic trial comparing the potential biosimilar tocilizumab (QX003S) with the reference product (Actemra ®) in Chinese healthy subjects. Ann Med. 2021;53(1):375–383.
- Subramanyam M. Immunogenicity of Biotherapeutics - An Overview. J Immunotoxicol. 2006;3(3):151–156.
- Burmester GR, Choy E, Kivitz A, et al. Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1078–1085.
- Benucci M, Meacci F, Grossi V, et al. Correlations between immunogenicity, drug levels, and disease activity in an Italian cohort of rheumatoid arthritis patients treated with tocilizumab. Biol: Targets Ther. 2016;10. 53–58.
- Hässler S, Bachelet D, Duhaze J, et al. Clinicogenomic factors of biotherapy immunogenicity in autoimmune disease: a prospective multicohort study of the ABIRISK consortium. PLoS Med. 2020;17(10):e1003348.
- Bray-French K, Hartman K, Steiner G, et al. Managing the Impact of Immunogenicity in an Era of Immunotherapy: from Bench to Bedside. J Pharm Sci. 2021;110(7):2575–2584.
- Schreitmüller T, Barton B, Zharkov A, et al. Comparative immunogenicity assessment of biosimilars. Future Oncol. 2019;15(3):319–329.
- Wang Y-MC, Wang J, Hon YY, et al. Evaluating and reporting the immunogenicity impacts for biological products - a clinical pharmacology perspective. AAPS J. 2016;18(2):395–403.
- Carrasco-Triguero M, Dere RC, Milojic-Blair M, et al. Immunogenicity of antibody-drug conjugates: observations across 8 molecules in 11 clinical trials. Bioanalysis. 2019;11(17):1555–1568.
- Roche Registration GmbH. RoActemra 20 mg/mL concentrate for solution for infusion. Summary of product characteristics. European public assessment report last updated 25/10/2022. European Medicines Agency. [Online]. [Cited 2022 Nov 2]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/roactemra
- Murthy S, Lee TCIL-6. blockade for COVID-19: a global scientific call to arms. The Lancet Respir Med. 2021;9(5):438–440.