
ABSTRACT
Background
Tocilizumab is a monoclonal immunoglobulin G interleukin-6 receptor antagonist. MSB11456 is a proposed tocilizumab biosimilar.
Objective
To determine the pharmacokinetic equivalence of a single subcutaneous injection of MSB11456, when delivered via autoinjector (AI) and prefilled syringe (PFS), in healthy adult subjects.
Research design and methods
In this randomized, open-label, single fixed-dose, crossover study, 91 subjects received subcutaneous administration of tocilizumab 162 mg via AI and PFS presentations. The primary endpoint pharmacokinetic parameters were analyzed using analysis of variance. Safety data were summarized descriptively.
Results
There were no differences in pharmacokinetic parameters between presentations, and safety parameters were comparable. The 90% confidence intervals for the geometric least squares mean ratios of all primary pharmacokinetic parameters were contained within the predefined 80.00% to 125.00% bioequivalence limits, indicating pharmacokinetic equivalence between the AI and PFS.
Conclusions
MSB11456 administration via AI was bioequivalent to administration via PFS. MSB11456 can be administered by AI or PFS, increasing the available range of self-injection devices.
Trial registration
The trial is registered at EudraCT, number 2020–003419-86.
Plain Language Summary
Tocilizumab is a biologic drug that is used to treat autoimmune diseases, including rheumatoid arthritis. MSB11456 has been shown to be equivalent to the US-licensed and EU-approved tocilizumab when administered by subcutaneous injection. There are different devices available to administer subcutaneous injections, and depending on the device, the patient’s experience can be enhanced, convenience and compliance increased, and cost-effectiveness ensured for patients taking this medicine. This randomized, single fixed-dose, crossover study tested the pharmacokinetic similarity of MSB11456 when given subcutaneously via an auto-injector device versus a pre-filled syringe device in 100 healthy subjects. A total of 91 healthy volunteers received MSB11456 via both auto-injector and pre-filled syringe using a crossover design. Blood was collected before the first dose and at regular intervals during the study to determine the pharmacokinetics of tocilizumab and ensure safety. This study found that the pharmacokinetics of tocilizumab following administration using the autoinjector and the prefilled syringe were equivalent, and the safety profiles were similar. These findings indicate that the auto-injector can be considered another option that can be used to subcutaneously inject MSB11456.
1. Introduction
Tocilizumab is a recombinant humanized monoclonal immunoglobulin G disease-modifying interleukin-6 (IL-6) receptor antagonist that is primarily used as an antirheumatic drug but also used to treat patients with COVID-19 [Citation1] and T cell-induced severe or life-threatening cytokine release syndrome [Citation2]. Tocilizumab binds specifically to both membrane-bound and soluble IL-6 receptors and has been shown to inhibit IL-6-mediated signaling through these receptors [Citation3]. This results in a non-linear, dose-dependent pharmacokinetic profile [Citation3,Citation4]. MSB11456 is a proposed tocilizumab biosimilar that has shown pharmacokinetic (PK) equivalence to US-licensed and EU-approved tocilizumab when administered subcutaneously to healthy adult subjects [Citation5]. The safety and tolerability profiles of MSB11456 and US-licensed and EU-approved tocilizumab have also been found to be similar [Citation5].
Auto-injector (AI) devices are currently marketed throughout the world, and evidence is emerging to suggest a patient preference for administration of tocilizumab subcutaneously [Citation6–8] and via AI devices [Citation9,Citation10]. An AI device has been developed for the MSB11456 subcutaneous (SC) formulation that can be self-administered conveniently by patients. This randomized, cross-over study (EudraCT Number 2020–003419-86) was to determine the PK equivalence of a single SC injection of MSB11456 via AI to MSB11456 via prefilled syringe (PFS) in healthy adult subjects.
2. Patients and methods
This was a randomized, open-label, single fixed-dose, two-treatment, two-period, crossover study in healthy male and female subjects conducted at two investigative sites in Poland (Biokinetica S.A., Phase 1 Unit in Jozefow and MTZ Clinical Research Sp. Z O O in Warsaw). Subjects who met all eligibility criteria during the screening period were admitted to the study site on day −1 and randomized to one of two treatment sequences (AI PFS or PFS AI) and one of three injection sites (lower abdomen, upper thigh, or outer area of the upper arm; in a 1:1:1:1:1:1 ratio to one of six treatment-sequence and administration-site groups); the same injection site was used in both periods. Randomization of the treatment sequences and injection sites used interactive response technology and was stratified by body weight category at baseline (≤80 kg and >80 kg). Subjects received SC administration of tocilizumab of 162 mg in a fixed volume of 0.9 mL on day 1 of each treatment period ().
Figure 1. Study design.
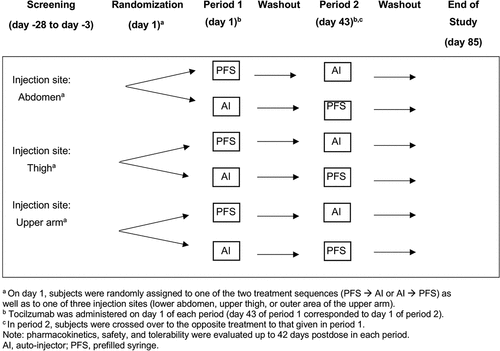
The study was conducted between 26 February 2021 and 12 June 2021, coinciding with the COVID-19 pandemic. Specific mitigation approaches were followed to protect subjects and ensure the integrity of data and minimize the risk of spreading severe acute respiratory syndrome 2 (SARS-CoV-2). Written informed consent was provided by all subjects before study entry, and the study followed the ethical principles of local regulations, the International Council for Harmonization guideline for Good Clinical Practice, and the Declaration of Helsinki.
2.1. Study population
Eligible subjects were healthy male or non-pregnant non-lactating female subjects, aged ≥18 to ≤55 years, with body weight between ≥60.0 to ≤100.0 kg, and body mass index (BMI) ≥18.5 to ≤30 kg/m2 at screening. Subjects were ineligible if they met any of the exclusion criteria including but not limited to history or presence of clinically significant atopic allergy or anaphylactic reactions; significant concurrent disease; unsuitable candidates as determined by the investigator teams; confirmed or suspected active COVID-19 infection; history of active or latent tuberculosis (as determined by QuantiFERON-TB Gold test), invasive systemic fungal infection, or serious infection (chronic or recurrent or frequent); previous exposition to tocilizumab or any other IL-6 acting drug; vigorous exercise within 72 hours of day 1; history of malignancy, diverticulosis, immunodeficiency, or hepatitis B; live vaccine within 12 weeks before the screening, or COVID-vaccine within 4 weeks before randomization.
2.2. Assessments
After treatment was administered on day 1 of each period, subjects remained confined at the study site until completion of the day 3 assessments. After discharge, subjects returned for a further 11 ambulatory visits at scheduled times in each period. A washout period of at least 42 days was scheduled between the two tocilizumab administrations, so that day 43 of period 1 corresponded to day 1 of period 2. A total of 31 samples for PK analyses were collected from each subject: each period pre-dose and at 8-, 12-, and 24-hours postdose, then regularly up until day 43 postdose. Safety assessments after day 1 included adverse event severity grading and causality assessment, laboratory assessments, vital signs, electrocardiograms, physical examination, and local tolerability. The severity of treatment-emergent adverse events (TEAEs) was graded according to the National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE) Version 5.0 (Grade 1: mild, Grade 2: moderate, Grade 3: severe or medically significant, Grade 4: life-threatening, and Grade 5: death related to AE).
2.3. Study endpoints
The primary objective of this study was to demonstrate the PK equivalence of PFS and AI presentations of MSB11456 after SC administration in healthy subjects. The secondary objective was to compare the safety and tolerability of PFS and AI presentations of MSB11456 in healthy subjects.
The choice of primary PK endpoints was in alignment with regulatory requirements (FDA, 2015): the area under the concentration–time curve (AUC) from time zero to the last quantifiable concentration (AUC0–last), maximum observed concentration (Cmax), and AUC from time zero to infinity (AUC0–inf). The parameters of time to Cmax (tmax), terminal elimination half-life (t1/2), and terminal elimination rate constant (λz) were assessed as secondary endpoints.
Secondary safety endpoints included TEAEs, predefined adverse events of special interest (AESIs) comprising serious or severe hypersensitivity reactions and serious infections, serious adverse events (SAEs), injection site reactions (ISRs), clinical laboratory values including hematology, chemistry, and urinalysis, vital signs, and 12-lead electrocardiogram (ECG).
2.4. Statistical analysis
Two data analysis sets were used, the PK Analysis Set used for PK evaluation and the Safety Analysis Set for drug safety evaluation. The PK Analysis Set included subjects who received the full dose of tocilizumab in both periods and who did not have any major protocol deviations or other events affecting PK assessment. The Safety Analysis set included all subjects who received at least one dose of tocilizumab.
Observed within-subject coefficient of variation (CV) for tocilizumab in the literature was up to 37.2% for AUC0-last and lower for Cmax and AUC0-inf [Citation10]. Assuming a departure of up to 5% from true bioequivalence and a CV of up to 38%, 80 evaluable subjects were needed to have at least 90% power for the 90% CI for each primary PK parameter comparison to be within 80.00% and 125.00%. Assuming no more than 20% subjects would be considered non-evaluable, 100 subjects were planned to be randomized.
Each primary parameter was analyzed with a linear mixed-effects analysis of variance (ANOVA) model on the natural logarithm of the endpoint, with treatment sequence (AI PFS or PFS AI), treatment period (1 or 2), treatment presentation (MSB11456 PFS or AI), baseline body weight strata (≤80 kg or >80 kg), administration site as fixed effects, and subject nested within the sequence as a random effect. Transformed back from the logarithmic scale, the geometric least-squares mean of the primary PK endpoints for each treatment presentation and the geometric least-squares mean ratio (GMR) (AI/PFS) with the corresponding 2-sided 90% confidence intervals (CI) were estimated.
PK equivalence between MSB11456 AI and PFS was demonstrated if the 2-sided 90% CI of the GMR was entirely contained within the predefined bioequivalence limits of 80.00% to 125.00% for all three primary PK endpoints. PK parameters were calculated using standard noncompartmental methods with the validated software Phoenix® WinNonlin® Version 8.1 (Pharsight Corporation, a Certara Company, Princeton, New Jersey, USA). All other statistical analyses were validated and performed using SAS Version 9.4.
Safety variables were summarized by treatment presentation and included numbers and percentages of subjects with TEAEs, SAEs, AESIs, local tolerability (including ISRs), vital signs, and clinical laboratory evaluations. Subject baseline demographics and characteristics were summarized descriptively.
3. Results
Of 253 subjects screened, 100 were randomized to one of the two sequences (AI PFS or PFS AI); all received at least one SC dose of MSB11456. Of these, 49 subjects were randomized to treatment sequence AI PFS and 51 to treatment sequence PFS AI. Overall, 97 subjects received MSB11456 AI and 94 received MSB11456 PFS; 91 subjects received both MSB11456 AI and PFS. Supplementary Figure 1 shows the patient disposition for the study. Demographic characteristics, including the baseline body weight categories (≤80 kg and >80 kg), were balanced across both treatment sequences (). A total of 38 (38.0%) female and 62 (62.0%) male subjects, aged between 18 and 54 years and with a baseline BMI between 20.0 and 30.0 kg/m2, participated in the study (). The baseline body weight ranged from 60.0 to 96.0 kg. Medical history data, concomitant medication usage, and treatment compliance were balanced across both treatment sequences. Subjects were injected in the lower abdomen (n = 34), the upper thigh (n = 33), and the upper arm (n = 33).
Table 1. Subject demographics and characteristics at baseline by treatment sequence (Safety Analysis Set).
3.1. Pharmacokinetics
3.1.1. Pharmacokinetic parameters of tocilizumab in serum
Following SC administration of 162 mg MSB11456 (via PFS or AI), quantifiable concentrations of tocilizumab were observed for most subjects 8 hours post-dose (first sampling time point). The overall shape of the arithmetic mean serum concentration–time profiles of tocilizumab was comparable between the two treatment groups. In both treatment groups, the tocilizumab concentrations reached a peak concentration at approximately 72 hours post-dose (); thereafter, tocilizumab concentrations decreased gradually by a nonlinear, biphasic elimination (a combination of linear clearance and Michaelis–Menten elimination).
Figure 2. Arithmetic mean (±SD) tocilizumab serum concentrationsa versus time on a linear scale following a single 162 mg subcutaneous dose of MSB11456 via auto-injector and prefilled syringe in healthy subjects (PK Analysis Set).
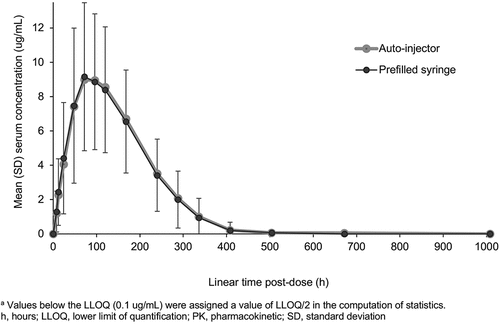
In the PK Analysis Set, exposure to tocilizumab was comparable between the AI and PFS presentations of MSB11456 in terms of all primary and secondary PK parameters (). When comparing the primary PK parameters of tocilizumab between the AI and PFS presentations, the GMR (90% CI) was 99.67% (90.95%, 109.21%) for Cmax, 102.88% (92.21%, 114.79%) for AUC0–last, and 100.23% (92.67%, 108.41%) for AUC0–inf (). The 90% CI for the GMRs of all three primary PK parameters were contained within the predefined 80.00% to 125.00% bioequivalence limits, indicating PK equivalence between the AI and PFS.
Table 2. Summary of tocilizumab serum PK parameters following a single 162 mg subcutaneous dose of MSB11456 administered via auto-injector and prefilled syringe presentation in healthy subjects (PK Analysis Set).
Table 3. Statistical analysis of the bioequivalence of a single 162 mg subcutaneous dose of MSB11456 administered via auto-injector and prefilled syringe in healthy subjects (PK Analysis Set).
Mean Cmax, AUC0–last, and AUC0–inf for tocilizumab were higher for subjects with a body weight ≤80.0 kg compared with >80.0 kg, after MSB11456 via both AI and PFS (). However, there was no significant difference in primary or secondary PK parameters between the two treatment groups within the body weight categories. There was also no difference in primary PK parameters between treatment groups when measured at the different injection sites ().
Table 4. Summary of tocilizumab serum PK parameters by baseline body weight category following a single 162 mg subcutaneous dose of MSB11456 administered via auto-injector and prefilled syringe presentation in healthy subjects (PK Analysis Set).
Table 5. Summary of tocilizumab serum PK parameters by injection site following a single 162 mg subcutaneous dose of MSB11456 administered via auto-injector and prefilled syringe presentation in healthy subjects (PK Analysis Set).
3.2. Safety
A total of 123 TEAEs were reported in 53 of 100 (53.0%) subjects. The safety profiles of a single SC dose of 162 mg MSB11456 administered via AI and PFS were comparable. About 16.5% of patients receiving the AI device and 18.1% of patients receiving the PFS device experienced a TEAE considered by the investigator to be related to the study drug (Figure 5). There was no distinguishing pattern of TEAEs, in terms of nature, frequency, or resolution, between the treatment groups (). Reported TEAEs were Grade 1 mild or Grade 2 moderate in severity for 46 (46.0%) subjects, Grade 3 for six (6.0%) subjects, and Grade 4 for one (1.0%) subject, who experienced an asymptomatic increase in blood creatinine kinase level to >10 times the upper limit of normal. Most Grade ≥3 TEAEs were identified in laboratory investigations, most commonly transient neutropenia, with no clinical signs or symptoms, and no notable imbalances between the treatment groups. No deaths were reported during this study.
Figure 3. The most commona treatment emergent adverse eventsb,c by preferred term following a single 162 mg subcutaneous dose of MSB11456 via auto-injector and prefilled syringe in healthy subjects (Safety Analysis Set).
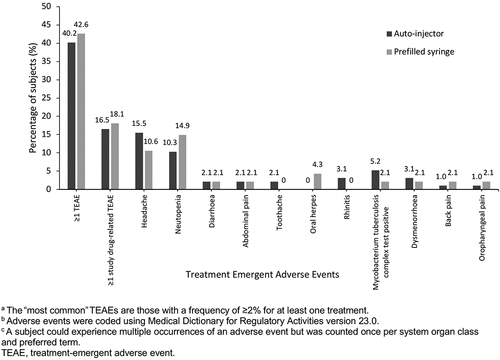
The only treatment-related TEAE reported by more than one subject was neutropenia (10.3% of AI recipients and 14.9% of PFS recipients). During this study, two (2.0%) subjects reported SAEs, specifically one SAE of SARS-CoV-2 test positive and one SAE of hypersensitivity. Both SAEs were reported as AESIs, and no other AESIs were reported during the study. The confirmed COVID-19 case was considered “otherwise medically important” and accordingly reported as a SAE. The proportion of subjects reporting at least one ISR was small, as expected (10 [10.0%] subjects overall), and comparable between the two devices. All ISRs were of Grade 1 to 2 severity. There were no clinically significant abnormal values or differences noted in vital signs, electrocardiograms, or physician examinations between the treatment groups.
4. Discussion
The primary objective of this study, to show PK equivalence between the MSB11456 AI and PFS devices after a single SC injection in healthy subjects, was met. In addition, secondary PK endpoints and safety parameters were comparable between MSB11456 AI and PFS. The serum concentration–time profile of MSB11456 was characterized by a short absorption/distribution phase, reaching a peak concentration at 72 hours post-dose, with a gradual decrease by biphasic elimination. This serum concentration trend was comparable between the two treatment groups. Consistent with a previous study of another biologic [Citation11], there was no significant difference between AI and PFS devices when tocilizumab PK exposure was stratified by body weight.
The best body site to inject SC biologics to enhance bioavailability is still a matter of debate. In general, rotating sites have been recommended. Some research reports unpredictability in the bioavailability between different administration sites (24% to 100% bioavailability) [Citation12], indicating high variability in site absorption and distribution from the interstitial tissue to the circulatory system [Citation13–16]. AI and PFS presentations did, however, show similar PK profiles for each injection site tested in this study.
At least one TEAE occurred in fewer than half the subjects after administration of either device (40.2% AI; 42.6% PFS). However, after administration using either device, less than one-fifth of patients (16.5% AI; 18.1% PFS) were found to have a TEAE related to the study drug. Many studies have reported similar proportions of subjects experiencing at least one adverse event with subcutaneous monoclonal antibody drug administration including tocilizumab (62% [Citation17], 53% [Citation11], 49% [Citation10], and 35% [Citation18]).
This study confirms that the PK of MSB11456 proposed tocilizumab biosimilar is equivalent for AI and PFS devices. Published research has also compared AI to PFS devices for alternative formulations of tocilizumab [Citation10] and other drugs like tumor necrosis factor inhibitors [Citation17] and monoclonal antibodies [Citation11,Citation18–22] and established PK equivalence, safety, and tolerability between devices. These studies found no statistical difference regardless of the device used [Citation10,Citation11,Citation17–22]. It is worth noting the potential benefits of AI administration, including convenience, ease of use (and consequently compliance), reduced risk of a dosage error, reduced anxiety from needle phobia, and integrated needle safety [Citation8–10,Citation23]. This study did not assess user preference or satisfaction with AI or PFS devices for the administration of tocilizumab. Only one study did not show any difference in usability preference between AI or PFS devices [Citation19], while many other studies reported high levels of user preference for AI over other methods of administration and advantages of time-saving, convenience, improved daily activities, and reduced cost [Citation6–10]. Further research into human factors could highlight the added benefits of AI devices for administration choice, flexibility, and convenience for potential at-home delivery.
Taken together, the results of this study demonstrate the PK equivalence of MSB11456 when administered by AI or PFS devices. MSB11456 was similarly well tolerated irrespective of which device was used by this study population of healthy adults. A potential limitation of the study is that it was open-label. However, this design was necessarybecause the two injection device presentations were visibly different. Although a double-dummy design could have been utilized, the increased burden on participants was not considered appropriate for a study with a primary objective of PK equivalence. The primary objective of evaluating PK equivalence was not expected to be influenced by the subject’s or investigator’s awareness of the treatment device used in each period and the bioanalytical laboratory that analyzed the tocilizumab serum PK samples were kept blinded to the randomization schedule. In addition, no immunogenicity analysis was planned per this study protocol, so the anti-drug antibody and neutralizing antibody status were not analyzed. However, no difference in immunogenicity with MSB11456 was expected based on delivery device. Immunogenicity data were collected in all other clinical studies of MSB11456 in which the reference product was used, namely in two PK studies comparing MSB11456 to the reference product following SC and intravenous routes of administrations and in a Phase 3 patient study.
5. Conclusion
This randomized open-label, single fixed-dose, cross-over study demonstrated PK equivalence between the AI and PFS presentations of MSB11456 after a single SC injection in healthy subjects. Furthermore, the primary and secondary PK parameters were overall comparable between the AI and PFS presentations across body weight categories and injection sites, as were the safety profiles. This study therefore supports the overall comparability of AI and PFS MSB11456 presentations, indicating that MSB11456 AI is an alternative delivery option to PFS that might be preferred by patients and increases the available range of self-injection devices.
Declaration of interest
M Ullmann, C Petit-Frere, J Monnet, and A Illes are employees of Fresenius Kabi SwissBioSim GmbH. C Dagres is a former Fresenius Kabi SwissBioSim GmbH employee. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Author contributions
M Tomaszewska-Kiecana, A Dryja, M Ullmann, E Vincent, C Petit-Frere, A Illes, C Dagres, J Monnet made substantial contributions to the conception, acquisition, design of the work, or analysis of interpretation of data. All authors were responsible for important intellectual content, critically revising the work, and making all content and editorial decisions. All authors had final approval of the manuscript version to be published and are accountable for all aspects of the work in ensuring the accuracy and integrity of this manuscript.
Research involving human participants: ethics approval
Before initiating the study, the investigator obtained written and dated approval from the Independent Ethics Committee (IEC) and regulatory authorities, for the study protocol and its amendments, written informed consent form (ICF), any ICF updates, subject recruitment material, subject information sheets, and other subject-facing material. Investigational New Drug (IND) (129,965) approval and EudraCT (2020-003419-86) registration were obtained.
This study was conducted in accordance with the Note for Guidance on Good Clinical Practice (GCP) International Council on Harmonisation (ICH) Harmonised Tripartite Guideline E6 (R2) from the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use ICH/135/1995 and requirements for the conduct of clinical studies as provided in the EU Directive 536/2014; the general guidelines indicated in the Declaration of Helsinki; and all applicable regulatory requirements.
Informed consent
Informed consent was obtained in compliance with the applicable regulatory requirements and adhered to GCP and to the ethical principles that have their origin in the Declaration of Helsinki. Each subject signed and dated two identical original ICFs, of which one was retained at the study site and the other was provided to the subject prior to participation.
As the study enrolled healthy volunteers and not patients, the medical benefit of the participants was limited to regular medical screenings including physical examination, measurement of vital signs, EGC, and safety laboratory assessments. Healthy volunteers also received financial benefit for their participation.
The trial is registered at EudraCT, number 2020-003419-86.
Supplemental Material
Download JPEG Image (387.6 KB){kind=link}
Acknowledgments
The sponsors acknowledge the participation of all healthy volunteers and the study site personnel involved in the clinical trial. Medical writing support was provided by Clare Koning and Caroline Spencer (Rx Communications, Mold, UK), funded by Fresenius Kabi SwissBioSim.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/1744666X.2023.2174970
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- World Health Organization (WHO). Prequalified Medicinal Products. Tocilizumab. Reference BT-CV001 (a). World Health Organization Prequalification Team (10 February 2022).
- Genentech. Actemra®(tocilizumab) injection, for intravenous or subcutaneous use. Highlights of prescribing information. 2022. South San Francisco (CA). [Online]. [cited 2023 Feb 6]. Available from: https://www.gene.com/download/pdf/actemra_prescribing.pdf
- Shetty A, Hanson R, Korsten P, et al. Tocilizumab in the treatment of rheumatoid arthritis and beyond. Drug Des Devel Ther. 2014;8:349–364.
- Navarro-Millán I, Singh JA, Curtis JR. Systematic review of tocilizumab for rheumatoid arthritis: a new biologic agent targeting the interleukin-6 receptor. Clin Ther. 2012 Apr;34(788–802.e3):788–802.e3.
- Schwabe C, Illes A, Ullmann M, et al. Pharmacokinetics and pharmacodynamics of a proposed tocilizumab biosimilar MSB11456 versus both the US-licensed and EU-approved products: a randomized, double-blind trial. Expert Rev Clin Immunol. 2022;18:533–543.
- Overton PM, Shalet N, Somers F, et al. Patient Preferences for Subcutaneous versus Intravenous Administration of Treatment for Chronic Immune System Disorders: a Systematic Review. Patient Prefer Adherence. 2021;15:811–834.
- Stoner KL, Harder H, Fallowfield LJ, et al. Intravenous versus Subcutaneous Drug Administration Which Do Patients Prefer? A Systematic Review. Patient 2014;8:145–153.
- Besada E. Potential patient benefit of a subcutaneous formulation of tocilizumab for the treatment of rheumatoid arthritis: a critical review. Patient Prefer Adherence. 2014;8:1051.
- Dashiell-Aje E, Harding G, Pascoe K, et al. Patient Evaluation of Satisfaction and Outcomes with an Autoinjector for Self-Administration of Subcutaneous Belimumab in Patients with Systemic Lupus Erythematosus. Patient. 2018;11(1):119–129.
- Fettner S, Mela C, Wildenhahn F, et al. Evidence of bioequivalence and positive patient user handling of a tocilizumab autoinjector. Expert Opin Drug Deliv. 2019;16(5): 551–561.
- Martin UJ, Fuhr R, Forte P, et al. Comparison of autoinjector with accessorized prefilled syringe for benralizumab pharmacokinetic exposure: AMES trial results. J Asthma. 2021;58(1): 93–101.
- Hu P, Wang J, Florian J, et al. Systematic Review of Device Parameters and Design of Studies Bridging Biologic-Device Combination Products Using Prefilled Syringes and Autoinjectors. AAPS J. 2020;22(2). DOI:10.1208/s12248-020-0433-8.
- Dua P, Hawkins E, Van Der Graaf P. A Tutorial on Target-Mediated Drug Disposition (TMDD) Models. CPT Pharmacometrics Syst Pharmacol. 2015;4(6):324–337.
- Smit C, De Hoogd S, Rjm B, et al. Obesity and drug pharmacology: a review of the influence of obesity on pharmacokinetic and pharmacodynamic parameters. Expert Opin Drug Metab Toxicol. 2018;14(3):275–285.
- Leung E, Crass RL, Jorgensen SCJ, et al. Pharmacokinetic/Pharmacodynamic Considerations of Alternate Dosing Strategies of Tocilizumab in COVID-19. Clin Pharmacokinet. 2022;61(2):155–165.
- Gradel AKJ, Porsgaard T, Lykkesfeldt J, et al. Factors Affecting the Absorption of Subcutaneously Administered Insulin: effect on Variability. J Diabetes Res. 2018;2018:1–17.
- Sabet A, Dickerson DS, Kunina EE, et al. A Randomised Controlled Trial Comparing the Pharmacokinetics and Tolerability of the Proposed Adalimumab Biosimilar MSB11022 Delivered via Autoinjector and Pre-filled Syringe in Healthy Subjects. Rheumatol Ther. 2022;9(2):693–704.
- Lon H-K, Cheng L, Nudurupati S, et al. Pharmacokinetic Comparability of Risankizumab Formulations in Prefilled Syringe and Auto-injector for Subcutaneous Injection. Clin Ther. 2021;43(3):629–636.
- Stauffer VL, Sides R, Lanteri-Minet M, et al. Comparison between prefilled syringe and autoinjector devices on patient-reported experiences and pharmacokinetics in galcanezumab studies. Patient Prefer Adherence. 2018;12:1785–1795.
- Ramael S, Moschetti V, Peter N, et al. AB0391 Similar pharmacokinetics, safety and tolerability of the Adalimumab biosimilar candidate BI 695501 administered subcutaneously via prefilled syringe (PFS) or autoinjector (AI) (Voltaire®-ai). BMJ. 2017;76:1185.
- Cherniakov I, Cohen‐Barak O, Tiver R, et al. A Pharmacokinetic Bioequivalence Study of Fremanezumab Administered Subcutaneously Using an Autoinjector and a Prefilled Syringe. Clin Pharmacol Drug Dev. 2021;10(9): 1018–1027.
- Cohen YZ, Zhang X, Xia B, et al. Pharmacokinetics of Subcutaneous Dupilumab Injection with an Autoinjector Device or Prefilled Syringe. Clin Pharmacol Drug Dev. 2022;11:675–681.
- Hupperts R, Becker V, Friedrich J, et al. Multiple sclerosis patients treated with intramuscular IFN-β-1a autoinjector in a real-world setting: prospective evaluation of treatment persistence, adherence, quality of life and satisfaction. Expert Opin Drug Deliv. 2015;12(1): 15–25.