
Abstract
The commercialisation of polymer electrolyte fuel cells (PEFCs) has been hampered by the high cost of platinum metal. Due to its high durability and catalytic activity, platinum is widely used to catalyse the oxygen reduction and hydrogen oxidation reactions essential to the operation of these cells. Core@shell nanoparticles with thin layers of platinum deposited on cores composed of cheaper materials have offered an attractive route towards the reduction of overall loading of platinum, with the retention of active catalyst surface area. This review surveys approaches taken to prepare idealised active and durable core@shell nanocatalysts by tweaking core compositions. A critical reflection on the current status of the field, as well as predictions as to likely directions for future developments, are discussed as a conclusion to the review.
Introduction
Nanoparticles are defined by the International Organization for Standardization (ISO) technical standard 80004-2 [Citation1] as materials for which all three of their external dimensions are in the nanoscale (1–100 nm). The ISO standard goes on to elaborate that materials in the nanoscale ‘often have properties that are not simple extrapolations of the properties of their larger form.’ That is to say, such nanomaterials exhibit physical and chemical properties not otherwise observed for macroscale objects of the same material. These unique properties have inspired significant scientific interest and research effort in a range of fields of human endeavour. Indeed, nanoparticles now find applications as diverse as in drug delivery [Citation2], chemical sensing [Citation3], and in protective ‘self-cleaning’ coatings [Citation4]. Another key application for nanoparticles is in catalysis, and particularly in electrocatalysis. In a recent review, Seh et al. highlighted the crucial role for electrocatalysis in securing a ‘clean energy’ future [Citation5]. In their work, the authors refer to one of the key lines of development in electrocatalysis being work to increase the number of (catalytically) active sites on a given electrode. One strategy towards doing so is the use of nanoparticulate catalysts, which by definition will have a greater number of active sites gram for gram compared to a bulk material, due to their high surface area to mass ratio. This is an intrinsic property of nanoparticles; due to their nanoscale dimensions, a greater proportion of their mass is exposed to the surface than an equivalent macroscale bulk material. With this in mind, nanoparticle (electro)catalysts have been the subject of significant research endeavour in recent years.
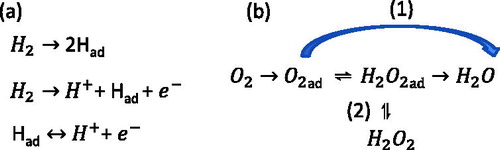
One particular application of nanoparticulate electrocatalysts is in catalysing the reactions required for the operation of low temperature fuel cells, such as the polymer electrolyte fuel cell (PEFC). These fuel cells utilise hydrogen fuel to generate electricity with water as the only by-product or emission, and may find applications in sectors including transportation and consumer electronics. A PEFC consists of an anode and cathode separated by a polymer membrane, typically composed of a fluorinated polymer such as Nafion®. At the anode, hydrogen fuel is dissociated in contact with the catalyst layer to produce two hydrogen (H+) ions, and two electrons (e−) in the hydrogen evolution reaction (HER). The mechanism of this is shown below, . It is generally accepted that the reaction proceeds along a three-step pathway, firstly involving adsorption of molecular hydrogen onto the catalyst surface. Once adsorbed, the molecule dissociates to liberate an ion and electron, alongside an adsorbed atom, which is further dissociated in a fast charge-transfer process [Citation6]. The liberated ions diffuse through the proton-conducting polymer membrane while the electrons are forced through an external circuit to generate electricity. At the cathode side of the PEFC, the electrons and protons recombine with oxygen ions (O−), generated by the catalytic reduction of molecular oxygen (O2) at the cathode catalyst, to produce water (H2O). The oxygen reduction reaction (ORR) proceeds via two potential pathways, shown in , which are often referred to as ‘direct’ and ‘indirect’. Direct oxygen reduction occurs in a four-electron pathway in which oxygen molecules adsorbed onto the catalyst surface are reduced directly to form water, without the formation of hydrogen peroxide (H2O2). The indirect pathway involves first the formation of adsorbed hydrogen peroxide, which either decomposes to re-form adsorbed oxygen, desorbs into the solution, or is further reduced to eventually form water [Citation6,Citation7]. In another fuel cell type, the direct methanol fuel cell (DMFC), the anode HER reaction is replaced with the methanol oxidation reaction (MOR), in which methanol is oxidised to liberate protons [Citation8].
Figure 1. (a) Three step mechanism for the hydrogen evolution reaction (HER). (b) The two known pathways for the oxygen reduction reaction (ORR), where (1) is the direct reaction and (2) is the indirect reaction.
Vignarooban and co-workers reflected on the ‘critical role’ that electrocatalysts play in determining the utility, durability and cost of PEFCs [Citation9]. The catalysts are typically composed of platinum (Pt) or other noble metals either dispersed directly on a carbon support or alloyed with other metals. As shown in , Pt, like other platinum group metals, is an expensive resource. Demand for Pt outstrips supply and the cost of the catalyst continues to limit the potential for fuel cells to become competitive with incumbent technologies. Physical approaches such as electrospraying [Citation10] and sputtering [Citation11] have been used to attempt to reduce platinum utilisation in PEFC electrodes with some success. However, the most significant research effort has been dedicated to developing novel nanoparticle electrocatalysts with reduced platinum loading, particularly through alloying.
Figure 2. Daily price per ounce for five platinum group metals graphed using data recorded between May 2015 and May 2018 by Johnson Matthey Precious Metals Management [Citation12]. Average per ounce prices in this period were: platinum $976.19, palladium $762.34, rhodium $1028.32, iridium $733.60 and ruthenium $76.40.
![Figure 2. Daily price per ounce for five platinum group metals graphed using data recorded between May 2015 and May 2018 by Johnson Matthey Precious Metals Management [Citation12]. Average per ounce prices in this period were: platinum $976.19, palladium $762.34, rhodium $1028.32, iridium $733.60 and ruthenium $76.40.](/cms/asset/5e06cf26-452b-45c5-a0ef-4893c7274930/tjen_a_1509383_f0002_c.jpg)
Alloying can increase the intrinsic activity of catalyst nanoparticles [Citation5], as well as influencing their durability. One category of alloy nanoparticles, specifically core@shell systems, has garnered significant attention from researchers. In these nanoparticles, a thin layer of one material is deposited on top of a core composed of another material. This category of nanoparticles is of particular interest in catalysis, as a layer of an expensive, catalytically active metal such as Pt can be layered on top of a core composed of a cheaper metal. The most obvious benefit of doing so is economic; a high surface area of active metal can be realised with lower mass of that metal. As is highlighted in a number of other reviews, however, there are catalyst issues other than cost impeding fuel cell commercialisation, including catalyst durability and activity [Citation7,Citation13–15]. The ideal core@shell catalyst system would confer economical savings while also realising enhanced durability and activity, however, and progress towards achieving these concurrent aims is the focus of this review.
Core@shell nanoparticle configuration
Element selection: d-block deliberations
Junliang Zhang and co-workers were among the first to report the use of core@shell nanoparticles to reduce platinum loading in fuel cell electrocatalysis. Using a metal deposition technique previously developed within the same group [Citation16], they used galvanic displacement to replace a copper adlayer with a monolayer of platinum on palladium (Pd) [Citation17], on alloy noble and non-noble metal cores [Citation18], and on a range of crystalline transition metal (TM) cores [Citation19]. They reported high activity relative to commercially available carbon-supported platinum (Pt/C) catalysts for their core@shell nanoparticles in ex-situ electrochemical testing, with reduced platinum loading.
In the intervening years, elements from all four corners of the TM block have been investigated as potential core materials. While Malacrida et al. [Citation20] prepared Pt-lanthanide alloys with Pt shells, and reported enhanced catalytic activity, most of the research effort has been focussed on the elements in the top corners of the d-block. Although describing alloys rather than core@shell particles, an oft-cited report which established the now ubiquitous ‘Volcano Plot’ () highlighted the high potential activity of Pt3Y [Citation21]. The volcano arranges Pt-based TM alloys according to literature-derived values for their kinetic current density, plotted against their computationally predicted oxygen adsorption energy. Knowledge of the oxygen adsorption energy of these nanoelectrocatalysts is crucial when attempting to predict their catalytic activity. The ideal ORR () catalyst will bind sufficiently strongly to oxygen to catalyse the reduction, but sufficiently weakly such as to encourage a high catalytic turnover.
Figure 3. Kinetic current densities (ln(jk/jkPt) vs. oxygen adsorption energies ΔEO − ΔEOPt (eV) for a range of bimetallic transition metal-platinum alloys. Reproduced from [Citation21] with permission from Springer Nature.
![Figure 3. Kinetic current densities (ln(jk/jkPt) vs. oxygen adsorption energies ΔEO − ΔEOPt (eV) for a range of bimetallic transition metal-platinum alloys. Reproduced from [Citation21] with permission from Springer Nature.](/cms/asset/eedf597b-9f31-40e1-83fc-351317747dbb/tjen_a_1509383_f0003_c.jpg)
In this study, it was observed that, as predicted, Pt3Y and Pt3Sc alloys kinetically outperformed Pt/C in electrochemical testing. Accordingly, the early TMs (Sc, Y, Ti) garnered significant attention from researchers looking to prepare highly active electrocatalysts – including those focussing on core@shell morphologies.
In no small part due to its low cost relative to Pt, Ti in particular has proven to be a popular candidate core/alloy material. In 2011, Jennings et al. [Citation22] highlighted the gulf between costs for the two elements – with approximate costs of £9.28 kg−1 for Ti and £3500.00 kg−1 for Pt in July 2011 – and examined Ti@Pt cluster ORR kinetics using density functional theory (DFT) calculations. Pure Pt and Ti clusters were studied, as were Pt32Ti6 and Pt6Ti32 configurations. The kinetic current densities and oxygen adsorption energies calculated for Pt3Ti, as shown on the volcano plot, , do not predict it to be the ideal catalyst [Citation21], however the authors of this study [Citation22] noted the variance in electronic properties expected of nanoparticle catalysts, relative to metallic bulk compositions of the same alloys. Indeed, the Pt–Ti clusters studied ‘exhibited favourable properties’ when compared to the pure Pt clusters. Namely, the Pt32Ti6 clusters bound hydroxyl groups less strongly than pure Pt clusters in simulation and this feature would be expected to confer enhanced ORR kinetics. Comprising 85% Pt and 15% Ti, though, it was conceeded that these clusters would be unlikely to offer significant economic advantages over the commercially available Pt/C catalysts [Citation22].
The authors additionally drew attention to propensity of Ti to form strong bonds with oxygen. Accordingly, Ti@Pt clusters have been experimentally elusive. Instead, Pt@TiO2 clusters have been prepared solvothermally [Citation23] and generated using a magnetron sputtering gas condensation cluster source [Citation24]. It appears that the TiO2 shell forms preferentially vis-à-vis the alternative Ti@Pt configuration, likely due to the affinity of Ti for oxygen. In the latter case the authors report slightly enhanced catalytic activity for their samples which contain Pt, relative to bare TiO2, but they did not benchmark these against commercial Pt/C. Given the synthetic challenges described, the search for the ideal core composition for ORR electrocatalysis must continue elsewhere.
Similar issues befell attempts to employ another cheap metal, chromium, as a core material in catalyst particles. Cr@Pt proved to be unattainable due to the formation of Cr oxides alongside pure Pt nanoparticles [Citation25]. Instead, the same group prepared non-segregated Pt–Cr alloys, which showed enhanced durability relative to Pt/C in electrochemical testing [Citation26].
Shifting focus towards the other end of the d-block, two interesting observations were made following studies of prepared Cu@Pt particles. Firstly, Cu@Pt nanoparticles with varying Pt shell thickness were prepared using ethylene glycol both as solvent and reducing agent [Citation27]. Although none of the particles demonstrated catalytic activity superior to that of commercial Pt/C catalysts, the authors noted a relationship between catalytic activity and shell thickness; the Cu@Pt particles with the thinnest Pt shells were the most catalytically active. In another work, Cu@Pt particles were prepared via sequential reduction of Cu and then Pt precursors on carbon. In initial electrochemical testing, the catalytic performance of the particles was similarly inferior to that of Pt/C, however the particles showed enhanced sustained activity over 1000 cycles [Citation28]. This durability enhancement requires further study.
In terms of achieving catalytic activity enhancements, a recent review of iron-containing Pt-based catalysts for fuel cell applications highlighted favourable weakening of Pt–O bonds, attributed to metal lattice shrinkage in Pt–Fe alloys [Citation29]. This effect was demonstrated experimentally in 2013 when Jang et al. [Citation30] prepared Fe@Pt nanoparticles with varying elemental composition via ultra-sound assisted polyol synthesis. In this study, Fe(acac)3 and Pt(acac)2 were mixed in a range of molarity ratios in ethylene glycol and sonicated. The particles formed were characterised using a range of microscopy and spectroscopy techniques, including X-ray diffraction (XRD). XRD was used to confirm the formation of a Pt shell on an Fe core due to the absence of characteristic iron or iron oxide peaks (). The researchers additionally performed a DFT analysis of oxygen binding with results suggesting weakened Pt–O bond formation for the formed particles. This expectation was confirmed by electrochemical measurements which showed that clusters with compositions Fe1.2@Pt (i.e. 55% Fe), with Pt monolayers were up to 6.5 times more active for the ORR than commercial Pt/C.
Figure 4. (a) X-ray diffraction patterns for a range of Fe@Pt samples showing clear shifted Pt peaks in the alloyed samples but no iron or iron oxide peaks and (b) a TEM image with sizing measurements on Fe@Pt nanoparticles. Reproduced from [Citation30] under Creative Commons licence (CC BY-NC 3.0)
![Figure 4. (a) X-ray diffraction patterns for a range of Fe@Pt samples showing clear shifted Pt peaks in the alloyed samples but no iron or iron oxide peaks and (b) a TEM image with sizing measurements on Fe@Pt nanoparticles. Reproduced from [Citation30] under Creative Commons licence (CC BY-NC 3.0)](/cms/asset/a04d29e0-2519-41b3-b461-8e570a8024ac/tjen_a_1509383_f0004_c.jpg)
Other work has focussed on incorporating nickel and cobalt into Ni@Pt, Co@Pt and indeed Ni–Co@Pt particles. Marking a step-change in approach, the synthesis of ordered core–shell nanoparticles with Pt3Co bimetallic cores coated by a 2–3 atom thick layer of Pt was reported in early 2013 [Citation31]. In this study, the researchers heat-treated a batch of their Pt3Co@Pt nanoparticles at 700 °C under a flow of H2 gas to engender ordering in the lattice structure of the Pt3Co core alloy. Physical and electrochemical characterisation results for an annealed sample, denoted Pt3Co/C-700 and an un-treated sample, Pt3Co/C-400 were compared. Annular dark field-scanning transmission electron microscopy (ADF-STEM) and electron energy loss spectroscopy (EELS) were used to map the elemental composition of the clusters and to confirm the formation of a Pt shell in each case. XRD measurements of the annealed samples highlighted increased lattice contraction and the pronouncement of (100) and (110) crystal phases, confirming the increased ordered crystallinity associated with this treatment. In initial electrochemical testing, both samples outperformed conventional Pt/C catalysts but of most interest is the trend identified after significant electrochemical cycling. After 5000 cycles, the electrochemically active surface area (ECSA) was measured for both samples, alongside further ‘post mortem’ physical characterisation. The authors noted that the electrochemical activity measured for Pt3Co/C-700 remained constant after these cycles, and EELS measurements and images confirmed that the nanoparticles remained well dispersed on the carbon supports. A 30% reduction in ECSA was observed for the Pt3Co/C-400 samples in the same timeframe. These results highlight the potential for improving catalyst durability through inducing molecular/crystalline ordering.
Ni@Pt particles have proven to be particularly popular catalyst candidates. Early work focused on the preparation of Ni@Pt particles with varying surface coverages of Pt through a modified polyol process [Citation32]. Catalysts with monolayer Pt shells were found to be the most active, with the researchers showing that increased coverage of Pt did not confer enhanced activity or durability.
Other researchers have exploited the difference in nobility between Pt and Ni to prepare ‘dealloyed’ catalysts. Corroding the catalysts either electrochemically, using potential cycling, or chemically, using acid, results in the selective removal of the less noble metal (in this case Ni) from the surface layers, producing particles with Pt-enriched surfaces. A recent review [Citation33] discussed the balancing act in dealloying, in which catalyst activity gains can be associated with losses in durability. Corrosive dealloying encourages sub-surface reorganisation of Ni which can engender optimised strain upon the Pt shell, thus enhancing catalytic activity. However, corroded particles can also exhibit porosity on the nanoscale, which makes them vulnerable to additional degrees and mechanisms of degradation. Studies [Citation34,Citation35] have shown that initial particle composition in terms of Pt:Ni ratio has a significant influence on eventual Pt shell thickness following dealloying. PtNi3 particles have shown the most significant enhancements in catalyst activity when compared to commercial Pt/C, relative to PtNi and PtNi5. This observation is made despite PtNi3 particles having the thickest Pt shells. These findings led the authors to elaborate on prior conclusions that catalytic activity scaled simply with shell thickness. Through aberration-corrected STEM imaging and EELS measurements (), the configuration of the particles was analysed. In particular, the researchers were interested in how platinum:nickel density varied across the profile of their particles and this is shown in the EELS line scans shown in panels b–d, f–h and j–l in . In the PtNi case (b–d) distinct core and shell components are observed with relatively smooth profiles. In the PtNi3 and PtNi5 particles (f–h and j–l, respectively), however, the line profiles show clear bumps, indicating areas of higher and lower concentration of Ni in the ‘core.’ The researchers thus describe the formation of Ni-enriched inner shells in their PtNi3 and PtNi5 particles. They surmise that enhanced strain is exerted upon the Pt outer shells by the inner Ni layers, enhancing catalytic activity, and that this effect is most significant in the PtNi3 system.
Figure 5. HAADF-STEM images and EELS line profiles of D-PtNi (a–d), D-PtNi3 (e–h) and D-PtNi5 (i–l) nanoparticles. Reprinted with permission from [Citation34]. Copyright © 2012 American Chemical Society.
![Figure 5. HAADF-STEM images and EELS line profiles of D-PtNi (a–d), D-PtNi3 (e–h) and D-PtNi5 (i–l) nanoparticles. Reprinted with permission from [Citation34]. Copyright © 2012 American Chemical Society.](/cms/asset/6aebde9f-c3d9-4e85-9eed-62c6a4ddf5e7/tjen_a_1509383_f0005_c.jpg)
A number of studies focussing on the preparation and characterisation of PtNiCo alloys, and NiCo@Pt particles have followed. Ternary alloys, and bimetallic alloys coated with Pt, demonstrating high catalytic activity and durability have been widely reported but the mechanism for these improvements is ill understood. In one particular work, Pt–Ni, Pt–Co and Pt–Ni–Co alloys were prepared, with thin Pt ‘skin’ outer layers deposited electrochemically [Citation36]. In their synthesis, the researchers first electro-deposited Ni or Co (or both Co and Ni in the Pt–Co–Ni case) onto a glassy carbon electrode surface. The coated electrodes were subsequently immersed in K2PtCl6 solution to permit replacement of Co and Ni with Pt, prior to a voltammetric dealloying step in which potential was applied across the −0.3–1.5 V range to form a stable Pt outer surface. ‘A few layers’ of Pt, with thickness of ‘a few nanometres,’ were deposited, so the authors suggest that total platinum loading was low in each case [Citation36]. These layers were characterised physically and electrochemically, and activity enhancements over Pt/C were observed in the range Pt–Ni–Co > Pt–Co > Pt–Ni. The authors concede that further work will be required to understand the origin of the degree to which ORR activity is enhanced for their ternary alloy, but they surmise that interactions between Co and Ni contribute additionally to electronic d-band effects and lattice constant mismatch such as to further weaken Pt–O bond strengths at the surface of their electrode.
Lattice mismatch and the associated surface strain are oft-cited, frequently poorly explained phenomena which are crucial to the development of ideal core–shell electrocatalysts. In further computational studies, Jennings and colleagues sought to understand these effects and predict which metal–platinum (M@Pt) combination would realise the theoretical optimum structure. In his doctoral research [Citation37], Jennings highlighted a triumvirate of properties which contribute to the stability of any intermetallic cluster structure – those being the surface area exposed, the surface energy of those external facets, and the internal strain associated with atomic rearrangements. These strain effects are particularly significant in clusters composed of metals of varying sizes and electronegativities. Larger atoms, and those which are more electronegative, are more inclined to adopt positions at the surface of a given cluster.
The size factor accounts for the mismatching of crystal lattices when shell layers of platinum form atop core surfaces with varying atomic radii. These strain contributors influence Pt–Pt bond length at the cluster surface, with knock-on impacts upon adsorbate binding energies in that shorter Pt–Pt bonding results in weaker Pt–O binding. These effects were examined and illustrated experimentally by Strasser and colleagues [Citation38] when they studied the constructive effect of compressive strain in Cu@Pt electrocatalytic clusters. In low energy electron diffraction experiments, the researchers observed a broadening of the electronic d-band of their Cu@Pt clusters and ascribed this to enhanced overlap between metal electronic clouds with decreasing atomic distances. The effect of this broadening contributes to enhance ORR kinetics at the surface of the particles prepared. To quantify the relationship between strain and catalytic activity, the researchers compared measured experimental values for their clusters to a theoretically derived volcano plot, shown in . Computational work suggested that the ORR activity would peak at a certain critical strain value, after which point Pt–O binding would be too weak for the reaction to occur readily. The researchers did not observe this trend, however, and suggest that this may be due to differences in strain experienced by Pt atoms at different positions within the particles. They hypothesise that Pt atoms adjacent to the Cu core will relax under strain to adopt lattice parameters closer to that of the Cu atoms, while surface shell Pt atoms – those directly engaging in catalysis – will relax towards lattice constants more typical for bulk Pt. Such an effect was described previously for Pt films grown on Cu (111) surfaces [Citation39]. These observations contributed significantly towards a more robust understanding of the theoretically predicted trends in core@shell catalyst activities, while also further demonstrating the complexity of these systems.
Figure 6. Experimentally verified ORR activities as a function of strain for Cu@Pt nanoparticles with red and blue triangles referring to particles annealed at 800 °C and 900 °C, respectively. The dashed line relates the theoretically predicted relationship. Reproduced from [Citation38] with permission from Springer Nature.
![Figure 6. Experimentally verified ORR activities as a function of strain for Cu@Pt nanoparticles with red and blue triangles referring to particles annealed at 800 °C and 900 °C, respectively. The dashed line relates the theoretically predicted relationship. Reproduced from [Citation38] with permission from Springer Nature.](/cms/asset/21b47166-dba2-428e-9014-2205e12374bc/tjen_a_1509383_f0006_c.jpg)
In a further theoretical work, Jennings et al. [Citation40] computationally analysed rates of oxygen dissociation at M@Pt surfaces relative to shell flexibility. This flexibility, intrinsically linked to lattice mismatch and core-size, permits the distorting of the shell to facilitate fast oxygen dissociation. These flexibility studies recommended alloying with late TMs (particularly Ni, Cu and Zn) as their interaction with platinum is sufficiently weak as to permit permutation of the outer shell layer. The authors conceded though that alloys and core@shell clusters composed of these elements were impeded by stability issues due to dealloying and dissolution. These findings all contribute to a growing understanding of the need to acknowledge and plan for factors additional to the d-electron band centre downshifting associated with core-metal electronegativity.
The structural and geometrical insights outlined thus far encourage conclusions which are at times rather contradictory. Rationally assessing all of the evidence available, it seems that M@Pt nanoparticles formed with either early or late TM cores have opposite advantages and disadvantages to recommend their use in ORR electrocatalysis. Where the early TMs, particularly Y, Sc and Ti have been lauded for their capacity to weaken Pt–O binding energies through their strong interactions with the Pt shell, so too have late d-metals such as Ni and Cu been recommended for their capacity to weakly interact with a thus highly flexible and attractively dissociative outer shell. Perhaps then the formula for the ideal M@Pt cluster requires a ‘just right’ compromise between characteristics associated with these two diverging poles. Cognisant of this observation or otherwise, a number of researchers have focussed their efforts on elements which adopt central positions within the TM block. Elements drawn from the ‘platinum group metals’, Pd, Rh, Ir, Ru and Os have thus piqued the interest of researchers and have been incorporated in core@shell cluster catalysts in a number of different conformations. Lu et al. [Citation41] prepared PtPd nanodendrites, while Pd@Pt nanocrystals were preferred by Qi et al. [Citation42]. Both groups reported enhanced catalytic activity ascribed to the formation of thin catalytic layers and ordered core–shell alignment, respectively. Ordered trimetallic core@shell catalysts have also been prepared. IrRe@Pt particles demonstrated comparable activity to commercial Pt/C with high durability [Citation43]. However, the major disadvantage associated with forming cores with the platinum group metals described is that they too are of limited abundance and useful in industrial processes. As a result, they are also expensive materials. A comparison of platinum group metal costs is shown in .
As shown, ruthenium, with an average price of 76.40 $/oz in 2015–2018, is an exception to the rule in that it comes in at least 12 times cheaper than platinum, which has averaged 976.19 $/oz over the same period. Accordingly, researchers have looked to ruthenium as a very eligible candidate for M@Pt studies. In 2013, ordered Ru@Pt nanoparticles were synthesised using a new method which reportedly minimised the formation of crystal lattice deformations. These nanoparticles were subsequently tested in a fuel cell stack to measure their capacity for catalysing the HER at a PEFC anode. Significantly, the researchers tested their nanoparticles with a carbon monoxide-poisoned hydrogen stream and were able to demonstrate enhanced tolerance to poisoning when compared to commercial Pt/C. This effect was ascribed in part at least due to the chemical ordering engendered in the Ru–core following an annealing step at 450 °C [Citation44]. This group once more highlighted the contribution that defined chemical ordering made to the activity and durability of their catalysts. This phenomenon has been further studied, with Cu@Pt–Ru nanoparticles tested as methanol and carbon monoxide oxidation catalysts in two successive works [Citation45,Citation46]. The authors noted in both cases that their nanoparticles demonstrated enhanced tolerance to carbon monoxide poisoning. The enhanced durability of each of the catalysts described highlights a unique property of alloyed Pt/Ru surfaces.
A more recent work with Ru@Pt HER catalysts sought to further investigate strain effects, and their impact upon the catalytic activity of Pt shells [Citation47]. To this end, the researchers prepared defined Ru@Pt particles alongside RuPt alloy catalysts. Higher hydrogen oxidation activities were reported for the Ru@Pt particles, with this attributed to weakened hydroxyl binding by the Pt shell as a result of high strain. The strain arises in the particles due to the mismatch between the platinum and ruthenium lattices.
Subsequent work with Ru@Pt clusters has focussed on oxygen reduction. One particular paper highlighted the potential to tune the ORR activity of the particles by varying the thickness of the Pt shell [Citation48]. Using an underpotential deposition technique, platinum mono- bi- and tri-layers were deposited onto a pre-prepared ruthenium core. These structures were confirmed using HAADF-STEM imaging and EELS measurements. In electrochemical testing, highest catalytic activities were observed for the Ru@Pt clusters with Pt bi-layer shells. The researchers sought to explain this finding using DFT calculations. These calculations show that for the catalysts studied (Ru@Pt1ML, Ru@Pt2ML, Ru@Pt3ML and Pt/C), Ru@Pt2ML struck the optimal balance in terms of fast O–O bond scission, but also in terms of facile hydrogenation in the removal of bound O.
A further paper described the preparation of core-shell like ‘Pt-surface-enriched’ Pt–Ir and Pt–Ru nanoparticles [Citation49]. In this work the researchers reported that their Pt–Ru nanoparticles achieved comparable catalytic activity to commercial Pt/C standards, but noted that these same nanoparticles performed very poorly in accelerated degradation testing, likely due to dissolution and dealloying.
Jackson and colleagues followed with recent work in which they prepared durable and active Ru@Pt catalysts using a wet chemical method [Citation50]. In their study, the researchers sought to evaluate the impact of varying nanoparticle precursor ratios, finding that the most active catalyst was that prepared with an Ru@Pt ratio of 1:1. Significantly, these optimised particles retained 85% of their activity following accelerated degradative cycling. The researchers reported that the Pt shell protected the otherwise vulnerable Ru core from corrosive degradation.
Incorporating non-metals
In order to address issues of durability, an additional class of core@shell materials have been investigated. In these systems, non-metal elements are incorporated into the alloy materials. In so doing, researchers have reported a range of novel properties.
Ternary compounds with metal nitride alloy cores were prepared by Kuttiyiel et al. [Citation51]. In this work, CoN@Pt, FeN@Pt and NiN@Pt nanoparticles were prepared and characterised electrochemically with respect to standard Pt/C. The increase in specific activity over Pt/C observed was ranked as follows: NiN@Pt > FeN@Pt > CoN@Pt. More significantly, in degradation testing all three catalysts were found to be more durable than Pt/C. Negative shifts in half-wave potential of 5 mV, 5 mV and 11 mV for NiN@Pt, CoN@Pt and FeN@Pt, respectively, which was compared to 40 mV for Pt/C. Aside from referring to the formation of nitrogen bonds, the authors do not offer any theories as to why nitriding the core metals gives rise to this effect. It seems reasonable to suggest that the metal nitrides would be less vulnerable to degradative oxidation than their pure metal equivalents given changes in their electronic configuration and bonding.
In a similar vein, researchers focussing on electrocatalysis for the MOR in DMFCs have observed that while Ru catalysts are more tolerant to methanol poisoning than Pt, they are vulnerable to oxidation. Several works have demonstrated the possibility to make these Ru catalysts more durable by forming alloys of ruthenium selenide, RuSe [Citation52,Citation53]. The mechanism for this improvement in durability has been ascribed to the formation of stable complexes between Ru and Se, which do not oxidise as readily as bare Ru metal.
Conclusions and outlook
Core@shell nanoparticles offer an attractive route towards decreasing platinum loading in electrocatalysts for fuel cells. As has been discussed, they also have the potential to realise enhanced catalytic activities and durabilities as compared to commercial Pt/C catalysts. This review has focussed on Pt–shell nanoparticles with a range of differing core materials, as this still reflects the approach taken by the majority of those publishing in this field. Indeed, as demonstrated, a wide array of candidate core materials have been studied by researchers, with virtually all of the TMs having been incorporated into TM@Pt assemblies. The resultant literature is at times dizzying given the sheer volume of systems being explored, and reticence within the community to adopt standardised testing procedures can make meaningful, quantitative comparisons between published works difficult. Several trends emerge from the works surveyed here, however as summarised in . The first is the initial attractiveness of candidate core materials drawn from the upper corners of the d-block. Cores composed of early TMs such as Sc, Ti and Cr were predicted to confer the most constructive electronic effects upon Pt shells, but these proved challenging to prepare given the electronegativities of the elements, and their propensities to readily oxidise and thus adopt shell positions [Citation24,Citation25]. Looking to the other end of the d-block, however, researchers have reported impressive relative activity gains for Co@Pt, Ni@Pt, and Cu@Pt particles, but these systems have been plagued with issues of poor durability [Citation54]. Despite their comparative expense, then, the platinum group metals have thus remained reliable candidates due to their nobilities. PGM@Pt particles are arguably more stable than the alternatives described but this stability arises in part due to a smaller electronic divergence from the bulk lattice constant or atomic radii of shell Pt atoms [Citation37,Citation40] than other elements described, so it is unlikely that these particles will contribute to significant, record-breaking gains in catalytic activity. Given the challenges associated with each group of TMs described, it is unlikely that ideal core@shell catalysts will be prepared simply by adopting the best possible candidate core material. Instead, it seems likely that researchers will seek to prepare optimised catalysts using a range of the materials described, focussing on addressing the issues highlighted with each candidate system through a range of other approaches. One standout observation from the works surveyed is the importance of – and challenges associated with – optimising shell thickness. While some researchers found that particles with thin (mostly monolayer) Pt shells were most active [Citation27,Citation32], others observed higher activities for particles with slightly thicker shells [Citation34,Citation35,Citation48]. Strasser et al.’s work with Cu@Pt highlighted the importance of balancing strain effects, which do not scale linearly with shell thickness, when designing catalysts [Citation38]. Other key priorities may focus on enhancing surface area and active site availability of catalysts; with one growing area of interest being the preparation of novel and ever more complex nano-morphologies, including nanowires [Citation55] and nanoframes [Citation56]. Meanwhile, we are sure to see a growing emphasis on developing novel strategies to enhance durability of some of the catalyst systems most prone to degradation. Ever more complex alloys with designed-in protection are one potential route, with third and fourth components, including p-block elements as per the nitriding example mentioned above [Citation51].
Table 1. Summarises key findings in terms of whether or not core@shell particles have been prepared for the given elements, and whether enhanced activities or durabilities have been reported for those particles. ‘Economy’ refers to expected cost savings or otherwise. Notes: (a) Refers to reference [Citation38]. (b) Notes that enhanced durability is reported but that the tests described are less rigorous than typical. (c) Notes that reported enhanced durabilities vary with annealing temperature.
Clearly it is difficult to choose any one leading candidate in this class of materials, and it is likely to become even more difficult to do so as these systems become ever more complex. Hopefully, though, researchers keeping the end application – and considerations of scale-up and commercialisation – will contribute to preparing better and better catalysts which can facilitate wider roll out of fuel cell technologies.
Additional information
Funding
References
- International Organization for Standardization. ISO/TS 80004-2:2015 [Internet]. Nanotechnologies – Vocab. – Part 2 Nano-objects. 2015 [cited 2018 Mar 26]. p. 10. Geneva, Switzerland: International Organization for Standardization. Available from: https://www.iso.org/standard/54440.html.
- De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3:133–149.
- Ionescu R, Cindemir U, Welearegay TG, et al. Fabrication of ultra-pure gold nanoparticles capped with dodecanethiol for Schottky-diode chemical gas sensing devices. Sensors Actuators, B Chem. 2017;239:455–461.
- Stieberova B, Zilka M, Ticha M, et al. Application of ZnO nanoparticles in a self-cleaning coating on a metal panel: an assessment of environmental benefits. ACS Sustain Chem Eng. 2017;5:2493–2500.
- Seh ZW, Kibsgaard J, Dickens CF, et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science. 2017;80:355.
- Markovic NM, Ross Jr. PN. Surface science studies of model fuel cell electrocatalysts. Surf Sci Rep. 2002;45:117–229.
- Rabis A, Rodriguez P, Schmidt TJ. Electrocatalysis for polymer electrolyte fuel cells: recent achievements and future challenges. ACS Catal. 2012;2:864–890.
- Wang M, Wang X, Chen M, et al. Nanostructured electrocatalytic materials and porous electrodes for direct methanol fuel cells. Chinese J Catal. 2016;37:1037–1048.
- Vignarooban K, Lin J, Arvay A, et al. Nano-electrocatalyst materials for low temperature fuel cells: a review. Chinese J. Catal. 2015;36:458–472.
- Martin S, Garcia-Ybarra PL, Castillo JL. High platinum utilization in ultra-low Pt loaded PEM fuel cell cathodes prepared by electrospraying. Int J Hydrogen Energy. 2010;35:10446–10451.
- Fabbri E, Taylor S, Rabis A, et al. The effect of platinum nanoparticle distribution on oxygen electroreduction activity and selectivity. ChemCatChem. 2014;6:1410–1418.
- Johnson Matthey Precious Metals Management. Price Charts - Platinum Group Metals [Internet]. Royston: Hertfordshire; 2016 [cited 2016 Mar 31]. Available from: http://www.platinum.matthey.com/prices/price-charts.
- Debe MK. Electrocatalyst approaches and challenges for automotive fuel cells. Nature. 2012;486:43–51.
- Shao M, Chang Q, Dodelet J-P, et al. Recent advances in electrocatalysts for oxygen reduction reaction. Chem Rev. 2016;116:3594–3657.
- Hwang SJ, Yoo SJ, Shin J, et al. Supported Core@shell electrocatalysts for fuel cells: close encounter with reality. Sci Rep. 2013;3:1309.
- Brankovic SR, Wang JX, Adžić RR. Metal monolayer deposition by replacement of metal adlayers on electrode surfaces. Surf Sci. 2001;474:173–179.
- Zhang J, Mo Y, Vukmirovic MB, et al. Platinum monolayer electrocatalysts for O2 reduction: Pt monolayer on Pd(111) and on carbon-supported Pd nanoparticles. J Phys Chem B. 2004;108:10955–10964.
- Zhang J, Lima FHB, Shao MH, et al. Platinum monolayer on nonnoble metal-noble metal core-shell nanoparticle electrocatalysts for O2 reduction. J Phys Chem B. 2005;109:22701–22704.
- Zhang J, Vukmirovic MB, Xu Y, et al. Controlling the catalytic activity of platinum-monolayer electrocatalysts for oxygen reduction with different substrates. Angew Chemie Int Ed. 2005;44:2132–2135.
- Malacrida P, Escudero-Escribano M, Verdaguer-Casadevall A, et al. Enhanced activity and stability of Pt–La and Pt–Ce alloys for oxygen electroreduction: the elucidation of the active surface phase. J Mater Chem A. 2014;2:4234.
- Greeley J, Stephens IEL, Bondarenko AS, et al. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat Chem. 2009;1:552–556.
- Jennings PC, Pollet BG, Johnston RL. Theoretical studies of Pt-Ti nanoparticles for potential use as PEMFC electrocatalysts. Phys Chem Phys. 2012;14:3134–3139.
- Wang P, Tooriyama H, Yokoyama K, et al. Smart decoration of mesoporous TiO2 nanospheres with noble metal alloy nanoparticles into core-shell, yolk-core-shell, and surface-dispersion morphologies. Eur J Inorg Chem. 2014;2014:4254–4257.
- Blackmore CE, Rees N V, Palmer RE. Modular construction of size-selected multiple-core Pt-TiO2 nanoclusters for electro-catalysis. Phys Chem Chem Phys. 2015;17:28005–28009.
- Gupta G, Iqbal P, Yin F, et al. Pt diffusion dynamics for the formation Cr-Pt core-shell nanoparticles. Langmuir. 2015;31:6917–6923.
- Gupta G, Sharma S, Mendes PM. Nafion-stabilised bimetallic Pt–Cr nanoparticles as electrocatalysts for proton exchange membrane fuel cells (PEMFCs). RSC Adv. 2016;6:82635–82643.
- Zhu H, Li X, Wang F. Synthesis and characterization of Cu@Pt/C core-shell structured catalysts for proton exchange membrane fuel cell. Int J Hydrogen Energy. 2011;36:9151–9154.
- Guterman VE, Belenov SV, Pakharev AY, et al. Pt-M/C (M = Cu, Ag) electrocatalysts with an inhomogeneous distribution of metals in the nanoparticles. Int J Hydrogen Energy. 2016;41:1609–1626.
- Antolini E. Iron-containing platinum-based catalysts as cathode and anode materials for low-temperature acidic fuel cells: a review. RSC Adv. 2016;6:3307–3325.
- Jang J-H, Lee E, Park J, et al. Rational syntheses of core-shell Fex@Pt nanoparticles for the study of electrocatalytic oxygen reduction reaction. Sci Rep. 2013;3:2872.
- Wang D, Xin HL, Hovden R, et al. Structurally ordered intermetallic platinum–cobalt core–shell nanoparticles with enhanced activity and stability as oxygen reduction electrocatalysts. Nat Mater. 2012;12:81–87.
- Chen Y, Liang Z, Yang F, et al. Ni-Pt core-shell nanoparticles as oxygen reduction electrocatalysts: effect of Pt shell coverage. J Phys Chem C. 2011;115:24073–24079.
- Strasser P, Kühl S. Dealloyed Pt-based core-shell oxygen reduction electrocatalysts. Nano Energy. 2016;29:166–177.
- Gan L, Heggen M, Rudi S, et al. Core-shell compositional fine structures of dealloyed Pt(x)Ni(1-x) nanoparticles and their impact on oxygen reduction catalysis. Nano Lett. 2012;12:5423–5430.
- Gan L, Cui C, Rudi S, et al. Core-shell and nanoporous particle architectures and their effect on the activity and stability of Pt ORR electrocatalysts. Top Catal. 2014;57:236–244.
- Li M, Lei Y, Sheng N, et al. Preparation of low-platinum-content platinum-nickel, platinum-cobalt binary alloy and platinum-nickel-cobalt ternary alloy catalysts for oxygen reduction reaction in polymer electrolyte fuel cells. J Power Sources. 2015;294:420–429.
- Jennings PC. Computational Studies of Mono- and Bimetallic Nanoclusters for Potential Polymer Electrolyte Fuel Cell Applications. Thesis, University of Birmingham; 2014.
- Strasser P, Koh S, Anniyev T, et al. Lattice-strain control of the activity in dealloyed core-shell fuel cell catalysts. Nat Chem. 2010;2:454–460.
- Fusy J, Menaucourt J, Alnot M, et al. Growth and reactivity of evaporated platinum films on Cu(111): a study by AES, RHEED and adsorption of carbon monoxide and xenon. Appl Surf Sci. 1996;93:211–220.
- Jennings PC, Aleksandrov HA, Neyman KM, et al. O2 dissociation on M@Pt core-shell particles for 3d, 4d, and 5d transition metals. J Phys Chem C. 2015;119:11031–11041.
- Lu Y, Du S, Steinberger-Wilckens R. Three-dimensional catalyst electrodes based on PtPd nanodendrites for oxygen reduction reaction in PEFC applications. Appl Catal B Environ. 2016;187:108–114.
- Qi K, Zheng W, Cui X. Supersaturation-controlled surface structure evolution of Pd@Pt core–shell nanocrystals: enhancement of the ORR activity at a sub-10 nm scale. Nanoscale. 2016;8:1698–1703.
- Karan HI, Sasaki K, Kuttiyiel K, et al. Catalytic activity of platinum monolayer on iridium and rhenium alloy nanoparticles for the oxygen reduction reaction. ACS Catal. 2012;2:817–824.
- Hsieh Y-C, Zhang Y, Su D, et al. Ordered bilayer ruthenium-platinum core-shell nanoparticles as carbon monoxide-tolerant fuel cell catalysts. Nat Commun. 2013;4:2466.
- Caballero-Manrique G, Velazquez-Palenzuela A, Brillas E, et al. Electrochemical synthesis and characterization of carbon-supported Pt and Pt-Ru nanoparticles with Cu cores for CO and methanol oxidation in polymer electrolyte fuel cells. Int J Hydrogen Energy. 2014;39:12859–12869.
- Caballero-Manrique G, Brillas E, Centellas F, et al. Electrochemical oxidation of the carbon support to synthesize Pt(Cu) and Pt-Ru(Cu) core-shell electrocatalysts for low-temperature fuel cells. Catalysts. 2015;5:815–837.
- Wang X, Zhu Y, Vasileff A, et al. Strain effect in bimetallic electrocatalysts in the hydrogen evolution reaction. ACS Energy Lett. 2018;3:1198–1204.
- Yang L, Vukmirovic MB, Su D, et al. Tuning the catalytic activity of Ru@Pt core-shell nanoparticles for the oxygen reduction reaction by varying the shell thickness. J Phys Chem C. 2013;117:1748–1753.
- Kaplan D, Goor M, Alon M, et al. Comparison of iridium- and ruthenium-based, Pt-surface-enriched, nanosize catalysts for the oxygen-reduction reaction. J Power Sources. 2016;306:219–225.
- Jackson A, Strickler A, Higgins D, et al. Engineering Ru@Pt core-shell catalysts for enhanced electrochemical oxygen reduction mass activity and stability. Nanomaterials. 2018;8:38.
- Kuttiyiel KA., Choi Y, Hwang S-M, et al. Enhancement of the oxygen reduction on nitride stabilized Pt-M (M = Fe, Co, and Ni) core–shell nanoparticle electrocatalysts. Nano Energy. 2015;13:442–449.
- Delacôte C, Lewera A, Pisarek M, et al. The effect of diluting ruthenium by iron in RuxSey catalyst for oxygen reduction. Electrochim Acta. 2010;55:7575–7580.
- Miecznikowski K, Kulesza PJ, Fiechter S. Application of black pearl carbon-supported WO3 nanostructures as hybrid carriers for electrocatalytic RuSex nanoparticles. Appl Surf Sci. 2011;257:8215–8222.
- Chen S, Gasteiger HA, Hayakawa K, et al. Platinum-alloy cathode catalyst degradation in proton exchange membrane fuel cells: nanometer-scale compositional and morphological changes. J Electrochem Soc. 2010;157:A82.
- Jiang K, Zhao D, Guo S, et al. Efficient oxygen reduction catalysis by subnanometer Pt alloy nanowires. Sci Adv. 2017;3:1–8.
- Niu Z, Becknell N, Yu Y, et al. Anisotropic phase segregation and migration of Pt in nanocrystals en route to nanoframe catalysts. Nat Mater. 2016;1:1–21.