
1. High performance computing in drug discovery
Drug discovery allows the discovery of molecules with pharmacological activity, and it is well-known to be a great time and cost consuming process. Indeed, there are millions of potential druglike compounds and the possibilities for de novo drug design are unlimited. This huge number of possible candidates has made it necessary to develop an automatic strategy to find the most appropriate compounds. Since the early nineties, concerted efforts from academic and commercial institutions have been made to build up a diversity of software able to analyze and predict key aspects in drug discovery such as protein – ligand interactions, pharmacophore and QSAR models, and Molecular Dynamics, among others. After 30 years of improvement, computational aided design has become a strongly consolidated strategy for drug discovery and design [Citation1,Citation2]. Currently, computational tools are being massively applied for predicting hit molecules against targets of interest, which is the approach called Virtual Screening (VS). However, the use of methods such as structural refinement, molecular docking or VS requires computing powers that only can be achieved by employing parallelized workflows, and even in those scenarios it is a bottleneck that limits advances in drug discovery. For example, in [Citation3], when seeking new chemotypes, a library of 138 millions of molecules was docked against the structure of dopamine receptor D4. About 70 trillion complexes were sampled requiring 43563 core hours or about 1.2 calendar days on 1500 cores. One should be aware that not all scientific users have access to these large supercomputing resources. In addition, this performance can be further improved by using advanced HPC architectures such as graphical processing units (GPUs). Hence, current computer aided drug design (CADD) has evolved from single-thread to multiple-thread architectures in order to increase computing power [Citation4,Citation5]. As an example, and show how GPU massive parallelization can drastically improve the time spent on a molecular docking task, depending on the depth of the search algorithm used [Citation6].
Figure 1. Execution time (in seconds) of six metaheuristic algorithms applied on a molecular docking task performed on a COMT dataset of DUD (http://dud.docking.org).
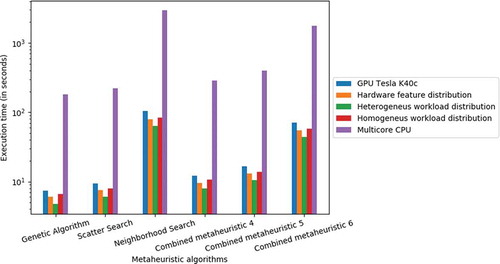
Table 1. Execution time (in seconds) of six metaheuristic algorithms applied on a molecular docking task performed on a COMT target dataset of DUD.
Also, due to the huge volume of data to be processed and the complexity of the calculations to be carried out by computational drug discovery (CDD) techniques, which mainly belong to the area of computational chemistry, the use of high performance computing (HPC) infrastructures in this context is a must nowadays. Basically, we can safely state that it would not be possible to process all the information available in the chemical databases of potential drug compounds, either accurately or quickly without HPC, since the required computational power is huge. In addition, HPC not only allows more data to be processed, but also in a shorter time. Regarding specific contexts, in outbreaks of infectious diseases where within a period of a week to a month they can become pandemic, HPC techniques can help to find a quick solution. For instance, in 2016 and due to the urgency of the Zika virus incidence in Brazil, Yuan et al. repurposed the old drug Novobiocin to treat such infection, with the main HPC based calculations only taking two days [Citation7]. Keep in mind that the development of a vaccine normally takes between 3 and 6 months, and the traditional process of developing new drugs may take up to 10 years [Citation8]. However, it needs to be mentioned that due to the complexity of modeling biological environments, some deficiencies remain unsolved, such as the premature discarding of druglike molecules or finding of PAINS (pan-assay interference compounds). Even so, nowadays we cannot begin to consider drug development without computational tools [Citation9].
From a practical perspective, we should highlight that drug discovery can greatly benefit from a diversity of HPC architectures, such as grid and cloud computing [Citation10], the latter being the most adequate solution for drug discovery calculations [Citation11]. Cloud computing is a very cost-effective solution to quickly access and exploit the required high computing power which allows academic institutions to perform their research with lower economic resources than large private companies. Indeed, cloud computing has enormously facilitated the development of CDD web servers, which are sometimes free of charge for the academic community [Citation12,Citation13]. They have become very popular due to their ability to process the information available in chemical databases by using a variety of software packages: In short, the usage of those servers and platforms has led to the development of new drugs and has promoted the emergence of novel techniques, methodologies and tools.
As part of all CDD approaches, we can underline the importance of VS, which has enormously facilitated the discovery of new drugs in the last two decades and decreased the previous costs in terms of time and money. In essence, VS methods possess the ability to screen large chemical databases aiming to extract small datasets made up of the most promising compounds, which are then prioritized for later stages. This strategy has shown to be much more efficient than its experimental counterpart in some cases, with high hit rate values [Citation14].
There are several topics related to VS techniques. Firstly, considering the large amount of information in databases to be processed, it is clear that the techniques do not go as deep as they should. This implies an apparent lack of knowledge that directly increases the number of false negatives and true positives, which are discovered in post in-vitro steps. However, VS techniques are valid regarding that traditional methods would not allow the same to de done. Besides, the nature of the techniques influences the final results. In search of maximum performance and a higher number of compounds processed per second, several approximations and reductions are made in the mathematical models. Also, algorithms used in certain techniques are designed to study performance so that they do not search for all the possibilities. This is counterproductive though. The time needed to analyze the compounds in the traditional way must first be taken into account. From now on, VS techniques must include as much information as necessary about the compounds, even if it involves an increase in time-consumption because It is important to note that this time will still be much shorter than if it were done traditionally.
However, and even with the aid of HPC, classical VS methods based mainly on physical and chemical principles have stagnated in terms of accuracy and ability to bring up novel chemotypes. In this limiting scenario, a new wave of HPC based VS methods has emerged to overcome the existing limitations; the explosion of Machine Learning (ML) and particularly Deep Learning (DL) methods in the last 5 years have reinforced the power of VS techniques and brought with it many new hopes. ML models must be previously trained with a dataset that contains compounds that will bind to the target and also non-binders. Applied to VS, ML techniques train models in such a way as to predict whether a potential inhibitor will bind to a target or its structure. In recent years, a large number of different models have emerged with impressive accuracy results [Citation15–Citation20]. Just to highlight some examples, Arús-Pous et al. used Recurrent Neural Networks [Citation21] to efficiently process a library of 975 million molecules, and Putin et al. developed and applied an original Deep Neural Network for the de novo design of novel small-molecule organic structures based on the Generative Adversarial Network paradigm and Reinforcement Learning [Citation22]. All these ML methods share common ground in that, instead of performing computationally expensive simulations or exhaustive Monte Carlo searches for all potential drug compounds from a large database, a properly trained ML model is capable of performing VS much faster than docking or Molecular Dynamics methods [Citation23]. However, we should be aware that ML methods are also accompanied by some practical limitations related mainly to the generally large dataset size they need for the training stage [Citation24]. In this respect, DL puts forward some approaches to overcome that problem. DL is a subfield of ML that uses a sequence of layers with non-linear processing units and learns high-level abstractions for data [Citation25]. In DL architectures, each layer trains for a distinct set of features based on the previous layer’s output. It makes DL networks capable of handling very large, high-dimensional data sets with billions of parameters that pass through nonlinear functions. Matrix-Matrix products are extensively used for neural network training and inferencing, so as to multiply input data and weights in the connected layers of the network. DL applications therefore work very well on computing systems that use accelerators such as GPUs or field-programmable gate arrays (FPGAs), which have been used in the field of HPC. This success has led to the appearance of processing chips specially designed for DL algorithms. For example, in 2016 Google announced that it had built a dedicated processor called the Tensor Processing Unit (TPU). Furthermore, NVIDIA is continuously improving its Tensor cores in order to accelerate matrix multiplication in the GPUs. IBM has also developed all the necessary software stacks for DL and AI applications such as PowerAI [Citation26]. Nowadays, the computing capacity of many supercomputing centers is divided into two totally different parts: a general purpose block and a block of these emerging ML and DL technologies.
A completely different emerging technology is quantum computing. Quantum computers take advantage of quantum mechanics to obtain features like parallel superposition, which allows them to efficiently compute parallel and distributed programs [Citation27]. Thanks to the nature of these quantum processes, solving fundamental problems for drug discovery related to modeling, simulation and molecular properties can be made possible. With the superposition of n qubits, the quantum version of a bit, 2 n possibilities can be represented and computed at the same time. That means many solutions can be explored at once, which makes it a strong candidate for solving optimization problems, such as finding the maximum similarity or the best docking. Currently D-Wave [Citation28] systems are already starting to use quantum annealing processes to find solutions to optimization problems. Companies such as IBM, ATOS, and GOOGLE are making concerted efforts to develop these new technologies and to provide access to prototypes or simulators of quantum computers to promote the development of quantum computing software, which provides an insight into future trends.
2. Conclusions
HPC is an essential part of the drug discovery process since it allows large quantities of data to be analyzed in a short time frame. The main advantage of HPC for drug discovery, and more specifically for VS, is the ability to discover novel molecules that would remain untapped without it. Accordingly, a currently important research line is related to the development of novel ML approaches in VS thanks to the exploitation of HPC resources, as opposed to more traditional approaches. Different solutions using DL or generative adversarial networks are achieving impressive results. Furthermore, due to the increasing interest of industry and academia in this sector, the development of ML/DL methods is accelerating very quickly. This is proven by the fact that academic institutions are working more and more on ML projects. This has been achieved thanks to the lower costs in computing hours either in physical clusters or on the cloud. In fact, the latter has experienced a considerable boost over the last decade, thanks to web servers that offer researchers the possibility of running experiments using different software and databases more economically.
3. Expert opinion
Some HPC based VS techniques are more accurate/advanced than others. The usual compound databases are so large that current VS methods cannot perform exhaustive searches. Instead, they mainly rely on local optimizers than can get easily stuck in local optima. Thus, they may discard promising compounds or even return erroneous recommendations. As an example we can consider rigid protein-ligand docking versus fully flexible Molecular Dynamics.
A clear role exists for HPC in CDD in the future. High-performance computing is a cross-disciplinary field that unites the modeling drug discovery processes with the use of parallel computers to extract valuable and reliable predictions from these models. As such, there is a clear need for multidisciplinary collaborations. Indeed, to achieve success in drug development, fruitful collaboration between the bio/chem communities (theoreticians, computational models and experimentalists) and specialists in the area of HPC computing and also experts from the different ML/Dl areas should take place. Thus, algorithms will not just be a mere tool, but an object of research themselves. Their efficient implementation will require understanding of computer architecture, parallel computing and specific abilities of software management.
In contrast with the private sector, Academia has very limited resources and it often happens that research projects involving medium to large scale experimental characterization are discarded because of their prohibitive costs. However, HPC based VS offers an opportunity to select the most promising druglike molecules from the whole database both quickly and cheaply [Citation29]. HPC should reduce its efforts to find new drugs and encourage academic institutions to move research toward this field. In addition, the price of HPC infrastructures and their maintenance cannot be afforded by all institutions, especially in academia. In this respect, the cloud computing services offered by companies such as Amazon, Google and Microsoft, are of vital importance. Furthermore, due to its huge potential, HPC is becoming a major driver for innovation offering possibilities that currently we cannot even evaluate or think about. Students and researchers employing these techniques are acquiring knowledge about high potential techniques and they are already prepared for upcoming needs in the next drug discovery projects.
It is important to mention that not all VS approaches need HPC resources, i.e. while large scale VS methods in the area of docking and pharmacophore have been applied routinely over the last 2 decades, we need to warn the reader that not all VS techniques need such HPC resources. In some areas of application of simple QSAR models, they can be easily trained and applied quickly on a regular desktop computer.
-Finally it should be noted that Moore’s law is coming to an end. As a consequence, scientists and engineers are focusing on innovative technologies, such as quantum computing. Quantum computers may well ensure the efficient provision of reliable results for many significant problems in quantum chemistry that cannot be solved by simulating processes in classical computation.
Declaration of interest
This research was partially supported by the supercomputing infrastructure at Poznan Supercomputing Center, the e-infrastructure program of the Research Council of Norway, the supercomputer center at UiT − the Arctic University of Norway and by the computing facilities at the Extremadura Research Centre for Advanced Technologies (CETA− CIEMAT), funded by the European Regional Development Fund (ERDF). CETA−CIEMAT belongs to CIEMAT and the Government of Spain. The authors also acknowledge the computing resources and technical support provided by the Plataforma Andaluza de Bioinformática at the University of Máaga. Powered@NLHPC research was partially supported by the supercomputing infrastructure at the NLHPC (ECM- 02). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Brogi S. Computational approaches for drug discovery. Molecules. 2019;24(17):3061.
- Schneider G, Clark DE. Automated De Novo drug design: are we nearly there yet? Angew. Chem Int Ed. 2019;58(32):10792–10803.
- Lyu J, Wang S, Balius TE, et al., Ultra-large library docking for discovering new chemotypes. Nature. 566(7743): 224–229. 2019.
- Samdani A, Vetrivel U. POAP: A GNU parallel based multithreaded pipeline of open babel and AutoDock suite for boosted high throughput virtual screening. Comput Biol Chem. 2018;74:39–48.
- Feinstein WP, Brylinski M. Accelerated structural bioinformatics for drug discovery. Boston: Morgan Kaufmann; 2015. DOI:10.1016/B978-0-12-803819-2.00012-4.
- Imbernón B, Cecilia JM, Pérez-Sánchez H, et al., METADOCK: A parallel metaheuristic schema for virtual screening methods. Int J High Perform Comput Appl. 32(6): 789–803. 2017.
- Yuan S, Chan JF, den-Haan H, et al. Structure-based discovery of clinically approved drugs as Zika virus NS2B-NS3 protease inhibitors that potently inhibit Zika virus infection in vitro and in vivo. Antiviral Res. 2017;145:33–43.
- Liu T, Lu D, Zhang H, et al. Applying high-performance computing in drug discovery and molecular simulation. Natl. 2016;3(1):49–63.
- Dantas RF, Evangelista TCS, Neves BJ, et al., Dealing with frequent hitters in drug discovery: a multidisciplinary view on the issue of filtering compounds on biological screenings. Expert Opin Drug Discov. 14(12): 1269–1282. 2019.
- Korb O, Finn PW, Jones G. The cloud and other new computational methods to improve molecular modelling. Expert Opin Drug Discov. 2014;9(10):1121–1131.
- Banegas-Luna AJ, Imbernón B, Llanes AC, et al. Advances in distributed computing with modern drug discovery. Expert Opin Drug Discov. 2019;14(1):9–22.
- Banegas-Luna AJ, Cerón-Carrasco JP, Puertas-Martín S, et al. BRUSELAS: HPC generic and customizable software architecture for 3D ligand-based virtual screening of large molecular databases. J Chem Inf Model. 2019;59(6):2805–2817.
- Panjkovich A, Daura X. PARS: a web server for the prediction of protein allosteric and regulatory sites. Bioinformatics. 2014;30(9):1314–1315.
- Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432(7019):862–865.
- Pereira JC, Caffarena ER, Dos Santos ER. Boosting docking-based virtual screening with deep learning. J Chem Inf Model. 2016;56(12):2495–2506.
- Wójcikowski M, Ballester PJ, Siedlecki P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci Rep. 2017;7(1):46710.
- Cang Z, Mu L, Wei G-W. Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening. PLoS Comput Biol. 2018;14(1):e1005929.
- Ragoza M, Hochuli J, Idrobo E, et al. Protein–ligand scoring with convolutional neural networks. J Chem Inf Model. 2017;57(4):942–957.
- Lima AN, Philot EA, Trossini GHG, et al. Use of machine learning approaches for novel drug discovery. Expert Opin Drug Discov. 2016;11(3):225–239.
- Lo Y-C, Rensi SE, Torng W, et al. Machine learning in chemoinformatics and drug discovery. Drug Discov Today. 2018;23(8):1538–1546.
- Arús-Pous J, Blaschke T, Ulander S, et al. Exploring the GDB-13 chemical space using deep generative models. J Cheminform. 2019;11(1):20.
- Putin E, Asadulaev A, Ivanenkov Y, et al. Reinforced adversarial neural computer for de novo molecular design. J Chem Inf Model. 2018;58(6):1194–1204.
- Carpenter KA, Cohen DS, Jarrell JT, et al. Deep learning and virtual drug screening. Future Med Chem. 2018;10(21):2557–2567.
- Lavecchia A. Machine-learning approaches in drug discovery: methods and applications. Drug Discov Today. 2015;20(3):318–331.
- Schmidhuber J. Deep learning in neural networks: an overview. Neural Netw. 2015;61:85–117.
- IBM PowerAI. Deep learning unleashed on IBM power systems servers. IBM RedBooks. Mar 2018. [online]. [cited 2020 Feb 17]. Available from: http://www.redbooks.ibm.com/abstracts/sg248409.html?Open
- Back R, Sere K Superposition refinement of parallel algorithms. In: Parker KR and Rose GA, editors. Formal description techniques, IV. North-Holland Publishing Company; 1992. p. 475–493.
- The mathematics of quantum-enabled applications on the D-wave quantum computer. Not Am Math Soc. 2019;66(6):1.
- Perez-Sanchez H, Wenzel W. Optimization methods for virtual screening on novel computational architectures. Curr Comput Aided-Drug Des. 2011;7(1):44–52.