
ABSTRACT
Introduction
G protein-coupled receptors (GPCRs) are the target of one-third of all approved drugs; however, these drugs only target about one-eighth of the human repertoire of GPCRs. GPCRs regulate a diverse range of critical physiological processes including organ development, cardiovascular function, mood, cognition, multicellularity, cellular motility, immune responses and sensation of light, taste, and odor. However, many GPCRs are expressed poorly, and a significant proportion have unknown ligands and unclear signaling pathways.
Areas covered
GPCRs are better suited to be targeted by monoclonal antibodies (mAbs) because of the challenges encountered in small-molecule discoveries such as druggability, selectivity, and distribution. mAbs have better drug-like properties in these respects. Herein, the authors review previously discovered functional mAbs that target GPCRs that are in the clinic and/or in development. They also review the biophysical considerations that make GPCRs so challenging to work with but also provide opportunities for biologic druggability.
Expert opinion
GPCRs are proven targets of small molecules yet remain an under-represented target of biologics. We believe that antibody drugs that target GPCRs have the potential to unlock new therapeutic avenues and also uncover previously unappreciated receptor biology, particularly when harnessing next-generation biologic modalities.
KEYWORDS:
1. Introduction
G protein-coupled receptors (GPCRs) have been targeted by many small molecules; however, antibodies and related biologics provide a unique opportunity for GPCR drug discovery. Here, we will review the molecular details of some of the described antibodies that target GPCRs and function as agonists and antagonists. In the second section, we will outline some of the challenges in GPCR drug discovery and describe how antibodies have unique properties to overcome these challenges.
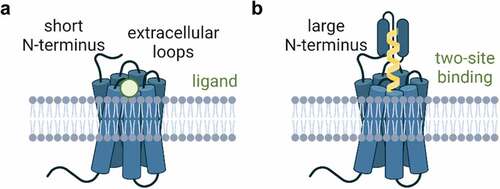
The human genome contains over 800 GPCRs, of which almost 400 are olfactory receptors. While olfactory receptor expression is mostly limited to the olfactory epithelium, some olfactory receptors are expressed in other tissues and targeting them may have therapeutic benefit [Citation1]. GPCRs are classified according to their sequence homology and phylogenetic analysis of their evolution [Citation2]. The Class A or rhodopsin-like family contains all of the olfactory receptors and many of the clinically targeted receptors including chemokine, amine, and peptide receptors. Class B1 receptors share a large extracellular N-terminus () and Class B2 belong to the adhesion GPCR family. Adhesion GPCRs are particularly intriguing antibody targets because of their unique extended extracellular domains [Citation3]. Class C GPCRs are thought to function as dimers, as are the Class T, taste receptors. Finally, the Class F receptors contain Frizzled and Smoothened, two important GPCRs that regulate organismal development.
Figure 1. G protein-coupled receptor extracellular topology and modes of ligand binding. A. model of short N-terminus found in many GPCRs. The short N-terminus and three extracellular loops make up all of the extracellular surface area of the receptor, and ligands bind deep within the transmembrane domains. B. Some GPCRs have large and extended N-termini with diverse functions. This model depicts a ligand that binds to the deep transmembrane domain ligand binding site as well as another binding site found in the large N-terminus. Created with BioRender.com.
Biological molecules (biologics) are a class of drugs that are produced by living cells [Citation4], for the purpose of this review, the biologics we will refer to most commonly are antibodies and their related fragments and bioactive peptides. Biologics have many unique properties that are not possible with small-molecule drugs. Many biologics take advantage of the properties of antibodies, in particular, the Fc (crystallizable fragment) region can be engineered to enhance drug half-life and induce antibody-dependent cellular cytotoxicity (or avoid cytotoxicity altogether) [Citation5]. Typically, antibodies diffuse through the blood stream, interstitial space, and lymphatic system and are excluded from the central nervous system. Unlike small molecules, antibodies are not cleared by the kidneys, but instead are degraded through Fc-receptor mediated endocytosis [Citation6]. Another advantage of biologics, as we will detail in this review, is that antibodies can be fused together to create bispecific molecules that target two different receptors, or biparatopic molecules that target two epitopes on the same receptor. Finally, because of their biological nature, it is possible to apply synthetic biology methods, such as directed evolution, to engineer antibodies, antibody fragments and peptides with desired drug-like properties [Citation7].
Despite the potential for novel therapeutic modalities, GPCRs have remained elusive for biologics drug development. Part of this is due to the structure of GPCRs in the plasma membrane. The extracellular surface of GPCRs follows a common topology (): an extracellular N-terminal domain and three extracellular loops [Citation8]. The extracellular loops of GPCRs tend to be very short, but can vary enormously in size from 5 to more than 170 amino acids [Citation9]. The orthosteric-binding pocket is where ligands bind and is located within the transmembrane domains. Most extracellular loops participate in ligand binding, although for small molecules these interactions only weakly contribute to ligand binding. Rhodopsin, the prototypical GPCR with highly detailed studies of activation states, has its orthosteric-binding pocket completely occluded by the extracellular loops and the ligand (retinal) diffuses laterally through the plasma membrane [Citation10].
Typically, GPCRs have short N-terminal domains, but many GPCRs also have extended N-terminal domains with a variety of functions (), including forming a second binding site for bitopic and allosteric ligands. The N-terminus of GPCRs is typically easy to produce as a soluble peptide and these peptides have frequently been used as antigens for antibody discovery campaigns. However, such antibodies would not be able to bind the extracellular loops. One of the biggest challenges in antibody-based GPCR biologics is to generate antibodies that actually bind to the orthosteric-binding pocket which is buried deep within the transmembrane domains. In the next section, we will discuss the mechanism of antibodies that bind to the N-terminus, extracellular loops, and orthosteric ligand-binding site and detail how that binding leads to activation or inhibition of the receptor ().
Table 1. Modality of G protein-coupled receptor targeted biologics. Summary of molecules discussed in Section 1: activators (agonists and positive allosteric modulators), blockers (antagonists, inverse agonists, and negative allosteric modulators), and next-generation molecules that target GPCRs. The disease indications of these biologics against GPCR targets, like small molecule therapeutics, span a broad range of indications from oncology to cardiometabolic disorders. Only three molecules are approved (dulaglutide, erenumab, and mogamulizumab) and only one molecule is in Phase 3 (talquetamab).
2. The functional activity of biologics targeted toward G protein-coupled receptors
2.1. Biologics that activate G protein-coupled receptors
The secretin family of Class B1 GPCRs has been targeted for agonist antibodies to treat diabetes and obesity. The clinically approved glucagon-like peptide 1 receptor (GLP-1 R) agonist dulaglutide (Trulicity) was engineered by Eli Lilly as a fusion of the GLP-1 peptide to the Fc (fragment crystallizable region) of IgG4 [Citation11]. IgG4 is known to engage with FcRn receptors to enhance antibody half-life via recycling [Citation12] and indeed, dulaglutide has a serum half-life of over 12 hours in humans, compared with less than 2–5 hours for exenatide and other GLP-1 peptide mimetics [Citation13]. Importantly, IgG4 does not activate the complement cascade which leads to antibody-dependent complement-dependent cytoxicity (ADCC) [Citation5]. Furthermore, the F234A and L235A mutations were introduced into dulaglutide’s IgG4 Fc region to reduce any possibility of ADCC [Citation11]. In summary, dulaglutide is a unique peptide agonist fused to an inert Fc region to activate GLP-1 R. In addition, Gmax Biopharm has developed glutazumab, a fusion of the GLP-1 peptide to the light chain of an anti-GLP-1 R antibody [Citation14]. Glutazumab is currently in clinical trials for the treatment of diabetes. Another such anti-GLP-1 R antibody fused to GLP-1 peptide to produce agonism, everestmab, has been described [Citation15]. Everestmab is a bispecific nanobody between an albumin-binding domain and a mutated GLP-1 peptide. Similarly, we recently described a GLP-1 peptide light-chain fusion to a GLP-1 R antibody called TB-059-002 [Citation16]. Amgen has recently reported positive data for a GLP-1 peptide fused to a glucose-dependent insulinotropic polypeptide receptor (GIPR) antibody, AMG-133, that functions as a dual GLP-1 R agonist/GIPR antagonist [Citation17,Citation18]. Tethering the GLP-1 peptide to the anti-GIPR antibody has the advantage of extending the half-life of the GLP-1 peptide and was shown to enhance localization of the molecule to the pancreas [Citation17]. AMG-133 is derived from a series of dual GLP-1 R agonists/GIPR antagonists and has recently been reported to show strong Phase 1 data in obesity. Ligand tethering with antibody delivery was also recently described for another Class B1 GPCR parathyroid hormone receptor (PTHR1) [Citation19].
Like GLP-1 R, GIPR and PTHR1, many GPCRs can have large, extended extracellular domains, typically at their N-terminus (). One GPCR, called thyrotropin receptor or thyroid stimulating hormone receptor (TSHR), has a large leucine-rich repeat extracellular N-terminal domain and autoantibodies have been discovered that activate TSHR by binding to this extracellular domain leading to Graves’ disease [Citation20–22]. Like TSHR, Class C metabotropic glutamate receptors (mGluR) have a large N-terminal domain with a Venus flytrap structure at the distal end, and camelid single-domain antibodies (nanobodies), DN10 and DN13, have been developed that activate mGluRs [Citation23]. The mGluR family of receptors require dimerization of the extracellular domains, and DN10 and DN13 both recognize unique epitopes that bridge the two dimerized extracellular domains. Interestingly, DN10 recognizes mGluR2 homodimers, while DN13 cross reacts with mGluR2 homodimers and mGluR2/mGluR4 heterodimers. This unique pharmacology was shown to be effective at enhancing the inhibitory action of mGluR2 agonists in neurons and DN13 impaired fear conditioning in mice. DN10 and DN13 were also recently used to detect mGluR2 homodimers and mGluR2/mGluR4 heterodimers in vivo using time-resolved FRET sensors labeling the nanobodies [Citation24]. In addition, a biased agonist antibody has been described which binds to the related receptor, mGluR7 [Citation25].
While these are examples of allosteric modulating antibodies that activate receptors through the large N-terminal extracellular domain, many GPCRs have a limited extracellular surface area, especially the Class A rhodopsin-like GPCR family (). Some antibodies have been shown to activate GPCRs by directly binding to the short extracellular loops and sometimes reaching into the orthosteric ligand-binding pocket. Amgen recently reported the design of a novel apelin receptor (APJR) agonist nanobody from a camel immunization with APJR solubilized and packaged into nanodiscs [Citation26]. The antibody, JN241, was found to block APJR activation, and the CDR3 extended into the orthosteric-binding pocket. Using modeling and molecular dynamics simulations, the authors converted JN241 into an agonist by introducing mutations into the CDR3 region. In addition, β1-adrenergic receptor (β1AR) antibodies were shown to be biased agonists, activating G protein but not β-arrestin signaling that were allosteric modulators [Citation27]. In fact, autoantibodies that activate β1AR have been implicated in dilated cardiomyopathy [Citation28–30]. Finally, β1AR antibodies have been described that activate the receptor in a similar way to autoantibodies [Citation31]. An X-ray free electron laser structure was determined for the serotonin 2B receptor (5-HT2B) bound to an antibody Fab fragment (P2C2) and the 5-HT2B agonist, ergotamine [Citation32]. P2C2 covers a total surface are of 945 Å2 of the extracellular loops of 5-HT2B, and almost all of the CDRs interact with the receptor. The HCDR3 of PC2C forms contacts with the extracellular loop 2 (ECL2) of 5-HT2B and also seems to interact with the plasma membrane. In this way, PC2C binds to the extracellular space of 5-HT2B, but not the orthosteric ligand-binding site, instead PC2C recognizes the active state of 5-HT2B. For the chemerin receptor (ChemR23), an agonist antibody was generated by using a peptide derived from the extracellular loop 3 [Citation33]. Many active-state specific antibodies have been described with robust functional data, including 5-HT4 [Citation34], the mu opioid receptor (MOR) [Citation35], delta opioid receptor (DOR) [Citation36], β2-adrenergic receptor (β2AR) [Citation37–40], muscarinic acetylcholine receptor 2 (M2) [Citation41], bradykinin receptor 2 (B2) [Citation42] sphingosine 1 phosphate 1 receptor (S1P1R) [Citation43], platelet activating factor receptor (PAFR) [Citation44] and chemokine receptor 5 (CCR5) [Citation45]. A summary of some of these activating molecules is presented in .
2.2. Biologics that block G protein-coupled receptors
Many antibodies have been described that act as G protein-coupled receptor antagonists. The secretin family B1 receptors have a large N-terminal domain that binds to an agonist that forms an alpha-helical rod (). We recently described a competitive antagonist antibody [Citation16] called TB-001-003 against GLP-1 R, which is a Class B1 or a secretin family GPCR. This antibody was derived from our GPCR-directed antibody library that has GPCR ligands grafted into an extended complementary determining region 3 (CDR3). Thus, TB-001-003 is able to displace GLP-1 from GLP-1 R, in a similar way to the peptide antagonist avexitide (exendin-(9-39)). In contrast, the GLP-1 R antagonist antibody, Fab 3F52, was raised against the GLP-1 R large N-terminus and was shown by x-ray crystallography to inhibit GLP-1 binding via an allosteric inhibition mechanism [Citation46]. Similar to TB-001-003, a GIPR antagonist antibody called Gipg013 was shown to displace the alpha-helical GIP peptide from the N-terminal domain of GIPR [Citation47]. Regeneron had advanced crotedumab [Citation48], another antagonist antibody for the secretin family GPCR called glucagon receptor (GCGR), to Phase 1 clinical trials for cardiometabolic disorders, but further development has been discontinued [Citation49]. Crotedumab was generated using a humanized mouse immunization of GCGR [Citation50]. REMD Biotherapeutics has advanced another GCGR antagonist antibody, volagidemab, to Phase 2 trials for the treatment of diabetes [Citation51,Citation52]. Finally, a nanobody antagonist was also developed against GCGR [Citation53]. Very few antibodies targeting GPCRs have been clinically approved globally; one is called erenumab to treat migraine. Erenumab binds to the calcitonin gene-related peptide type 1 receptor (CALRL) complexed to a receptor activity-modifying protein 1 (RAMP1) [Citation54]. The complex of CALRL and RAMP1 is called calcitonin gene-related peptide receptor (CGRPR) and erenumab was raised against an asymmetric Fc fusion of the RAMP1 and CALRL extracellular domains [Citation55,Citation56].
Other receptors with large N-terminal domains have been targeted with antibody antagonists. OncoMed reported vantictumab blocks the Frizzled family of receptors (FZD) which has cross-reactive antagonist activity of FZD1, FZD2, FZD5, FZD7, and FZD8 [Citation57] and blocks Wnt signaling for antitumor activity. FZD receptors possess a large N-terminus that contains a cysteine-rich domain at the distal end. The leucine-rich repeat GPCR (LGR) family enhances Wnt signaling through FZD, has a large N-terminus and LGR5 has also been targeted with the antagonist antibody called KM4056 [Citation58] developed by Kyowa Kirin.
A number of antagonist antibodies have been described that target the Class A rhodopsin-like GPCR family. The FDA granted IND clearance to getagozumab, an endothelin receptor A (ETA) antagonist antibody [Citation59] and another ETA antagonist antibody called AG8 was also recently characterized [Citation60]. MedImmune have described the discovery and optimization of Fpro0165, a formyl-peptide receptor 1 (FPR1) antagonist antibody that was shown to bind to the extracellular loop 2 of the receptor by docking the antibody to a structure of the receptor [Citation61]. Additionally, a series of synthetic nanobodies, including the optimized antibody called AT118i4, have been described that function as antagonists of the angiotensin II type 1 receptor (AT1R) [Citation62,Citation63]. Ribosome display from a library of mice immunized with a fragment of the cholecystokinin-B receptor (CCK-BR) extracellular domain was used to discover an effective antagonist antibody called scFv 77–2. scFv 77–2 was effective in preclinical models of neuropathic pain [Citation64]. Similarly, immunization with the N-terminus of the melanocortin-4 receptor (M4R) produced an inverse agonist antibody [Citation65]. Interestingly, a negative allosteric modulator of the β1AR was shown to bind to the extracellular loop 2 region of the receptor using sequence differences between the human and Turkey receptors [Citation66]. An antagonist nanobody called CA4910 was described for ChemR23 that showed enhanced antagonist efficacy when reformatted into a biparatopic molecule [Citation67]. An N-terminal peptide was used to generate a kappa-opioid receptor (KOR) antagonist antibody [Citation68]. Goldfinch Bio has advanced GFB-024, a cannabinoid-1 receptor (CB1) inverse agonist into clinical trials for obesity [Citation69,Citation70]. The complement 5a receptor 1 (C5AR1) is implicated in inflammatory diseases and cancer and Innate Immunity has advanced avdoralimab, a C5AR1 antagonist antibody, through the clinic for both of these indications [Citation71]. C5AR1 antagonist antibodies have also been shown to be effective in cardiometabolic disorders [Citation72]. In addition, C5AR1 antagonist antibodies have been discovered using the N-terminal peptide as an antigen for synthetic phage-display libraries [Citation73] and N-terminal peptide binding was determined to be the epitope for other C5AR1 antagonist antibodies [Citation74,Citation75].
GPCR antagonist antibodies have been heavily developed for chemokine receptors (called CCR and CXCR) in order to modulate the immune system in different disease states. Chemokine receptors belong to the Class A rhodopsin-like family of receptors with limited extracellular surface area. Mogamulizumab is a clinically approved CCR4 antibody [Citation76] for leukemia, but its mechanism of action is to induce antigen dependent cellular cytotoxicity (ADCC) killing of the tumor cells, rather than direct antagonism of CCR4, which has also been described for other CCR4-targeting antibodies [Citation77]. Another CCR4 antagonist antibody has been described that blocks CCR4 function [Citation78] and was later delivered to tumors via adeno-associated viral mediated expression of the antibody [Citation79]. Antibody antagonists have been intensely developed for the C-X-C chemokine receptor 4 (CXCR4) to treat cancer, including LY2624587, a potent antagonist [Citation80,Citation81]. In addition, ulocuplumab was developed and shown to be a competitive antagonist for the CXCR4 endogenous ligand, CXCL12 [Citation82]. Other CXCR4 antibodies have been targeted to and shown to compete for CXCL12 in the orthosteric-binding pocket [Citation83] and extracellular loops [Citation84–87]. CXCL12 binds to CXCR4 and also CXCR7, for which an inhibitory nanobody has been described [Citation88]. CXCR2 is another important chemokine receptor that is dysregulated in cancer. A novel biparatopic antibody was described that combined two nanobodies with weak efficacy, which resulted in a strong inverse agonist molecule [Citation89]. In addition, monoclonal antibodies have been described against CXCR2 using phage-display synthetic antibody libraries [Citation90]. CCR5 is a well-validated co-receptor for HIV-1 entry, and a plethora of antibodies have been developed that block CCR5 signaling and HIV-1 binding [Citation91–95]. Finally, antibody antagonists have been described for CCR2 [Citation96,Citation97], CCR7 [Citation98], and CCR9 [Citation99]. Viral GPCRs have also emerged as therapeutic targets of interest, such as the viral chemokine receptor encoded by cytomegalovirus called US28. Overexpression of US28 has been implicated in glioblastoma and antagonist antibodies have been described for US28 for the potential treatment of glioblastoma [Citation100,Citation101]. The antagonists, inverse agonists, and negative allosteric modulators described in this section are summarized in under ‘blockers.’
2.3. Next generation biologics that target G protein-coupled receptors
Next-generation antibody therapeutics have advanced with complex and elaborate structures and mechanisms of action [Citation102] and a significant proportion of GPCR-targeting biologics encompass these next-generation modalities.
Mogamulizumab is a clinically approved CCR4 antibody [Citation76] for leukemia. Kyowa Kirin first developed mogamulizumab and its mechanism of action is to induce ADCC, which depletes CCR4-expressing cells [Citation103]. Mogamulizumab was engineered to have reduced fucosylation, which enhances ADCC [Citation104]. Similar to CCR4, CCR8 is being targeted for ADCC depletion, as CCR8 is expressed specifically on immunosuppressive regulatory T cells (Tregs) in the tumor microenvironment. A novel tetravalent nanobody called Nb-Fc1b was targeted to CCR8-expressing Tregs and inhibited tumor growth [Citation105]. Gilead has advanced GS-1811 to Phase 1 trials in solid tumors to trigger ADCC in CCR8 expressing Tregs [Citation106]. Similarly, Bristol-Myers Squibb has an anti-CCR8 antibody called BMS-986340 in Phase 1 trials in solid tumors [Citation107]. However, as demonstrated in CCR8 knockout mice, the targeting of CCR8 requires ADCC to eliminate Tregs, as blocking the signaling of CCR8 has no effect on Treg mediated immunosuppression [Citation108].
Many molecules make use of bispecific binding, where each arm of the antibody binds to different antigens. An LGR5 and epidermal growth factor receptor (EGFR) bispecific antibody called MCLA-158 was recently discovered by screening a library of bispecific antibodies in patient-derived organoids [Citation109]. BsNb PX4 is a bispecific antibody composed of two nanobodies, one that targets CXCR4 and another targeting PD-L1 [Citation110]. BsNb PX4 is more effective than treating mouse models with each nanobody separately in a tumor type that is resistant to PD-L1 inhibition. Talquetamab is a bispecific antibody in Phase 3 clinical trials for multiple myeloma that targets CD3 and GPCR Class C, group 5, member D (GPRC5D) which is highly expressed on multiple myeloma cells [Citation111]. Additionally, a CXCR5 and CD3 bispecific antibody has been described [Citation112].
Antibody-drug conjugates (ADC) have been developed for many GPCR targets. For CXCR4, unnatural amino acid incorporation was used to introduce surface residues into an anti-CXCR4 antibody, which allowed conjugation with auristatin, an antimitotic agent [Citation113] for the treatment of cancer. This strategy was later improved upon for the antibody called ADC 713 by increasing the drug-to-antibody ratio (DAR) which resulted in lower toxicity [Citation114]. In addition, an Lck inhibitor called dasatinib was conjugated to a CXCR4 antibody in order to suppress T cell function in autoimmune disorders (as Lck expression is restricted to T cells) [Citation115]. Finally, a molecule called 4M5.3X4 was described that is a small interfering RNA against an HIV protein conjugated to a CXCR4 nanobody to suppress HIV replication in CXCR4 positive T lymphocytes [Citation116]. The viral GPCR US28 has been targeted by a molecule called VUN100-PS, which is a nanobody conjugated to a photosensitive drug to kill tumors expressing this receptor [Citation101].
Chimeric antigen receptor (CAR) T cell therapy is an advanced form of cellular reprogramming to target immune cells to tumors in order to treat cancer. The orphan GPRC5D has been shown to be overexpressed in myeloma cancer cells and has been targeted for a chimeric-antigen T (CAR-T) cell therapy called MCARH109 [Citation117]. Similar to CAR-T targeting, an oncolytic herpesvirus (oHSV) was engineered to target CXCR4+ glioblastoma cells by fusing the virus to a CXCR4 nanobody [Citation118] called oHSV/Nb-gD. oHSV/Nb-gD was also made to induce the expression of TRAIL, a TNF family ligand that induced apoptosis in CXCR4 positive glioblastoma cells to reduce tumor growth. Finally, the adhesion GPCR called brain angiogenesis inhibitor 1 (BAI1) was used to engineer a chimeric receptor for efferocytosis (CHEF) called BELMO that increased phagocytosis and alleviated inflammation in vivo [Citation119].
3. Opportunities and challenges in G protein-coupled receptor biologics discovery
3.1. Many G protein-coupled receptors are orphans
Approximately 30% of the ~400 non-olfactory GPCRs are orphans, with no known ligand and poorly understood signal transduction properties [Citation120]. As discussed above, many antibodies directed against GPCRs function as either allosteric or orthosteric activators. Thus, antibody ligands could be used de-orphanize GPCR signaling pathways, using technologies such as TRUPATH [Citation121], APEX [Citation122,Citation123], or PRESTO-TANGO [Citation124]. For example, many molecules target GPRC5D for CAR T cells, as GPRC5D is highly expressed in myeloma cancer cells [Citation117]. However, little is currently known about the ligands or signal transduction mechanisms for GPRC5D [Citation125]. So, GPRC5D antibodies could be used to profile signaling through APEX labeling, which would then provide a screening platform for deorphanization of endogenous ligands.
3.2. How synthetic biology enables G protein-coupled receptor biased agonists
GPCRs are able to transmit pleiotropic signals, through various G protein family members and β-arrestins and in this scenario it may be possible to target beneficial pathways with so-called biased agonists [Citation126]. As was demonstrated with the APJR antibody JN241-9 [Citation26], it is possible to change the functional characteristics of an antibody ligand by making point mutations (in that case an antagonist was converted to an agonist). Thus, mutagenesis can be used as a platform for structure-based drug discovery of biologics. Furthermore, with parallel testing of each GPCR signaling pathway, it may be possible to capture biased antibody ligands that signal through a therapeutically beneficial pathway and avoid side effects [Citation127]. The rapid and scalable synthesis of DNA has enabled the development of robust and diverse antibody libraries [Citation128], which allows for the discovery of antibodies that can bind to cryptic sites in GPCRs, such as the orthosteric ligand-binding pocket (). DNA synthesis also allows for large-scale mutagenesis of orthosteric ligand-binding antibodies, which would be a strategy to design biased agonist antibodies against GPCRs. In addition, nanobodies that bind to the intracellular side of the receptors can be used to stabilized different receptor conformations, which can accelerate drug discovery strategies [Citation129].
Figure 2. Opportunities and challenges in G protein-coupled receptor biologics discovery. A. Antibodies can target the ligand binding (orthosteric) site or other allosteric regions of GPCRs. B. Antibodies can stabilize GPCR conformations, allowing for complex pharmacology (positive and negative modulators, inverse agonists, etc) C. Antibodies can activate the diverse signals downstream of GPCRs and be engineered to selectively activate certain pathways. D. Antibodies can disrupt protein interactions in the same cell (trans) or in cis (in different cells) including blocking higher order GPCR oligomer formation. Created with BioRender.com.
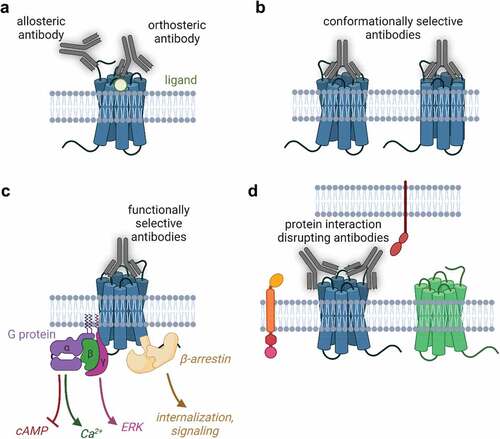
We have detailed the molecular mechanism by which some antibodies affect GPCR signaling and classified them into two broad categories: orthosteric or allosteric binders (). In addition, these antibodies are capable of stabilizing GPCR conformations () which is one way to drive functional selectivity downstream of the receptors ().
3.3. Cell-type specific expression of G protein-coupled receptors
As drugs, antibodies have the unique property of cell-type specific targeting. Rather than acting systemically and undergoing immediate metabolism, antibodies tend to accumulate in tissues with high concentrations of antigen. GPCRs are often expressed at very low levels and in specific tissue types. Large-scale datasets, such as the human protein atlas [Citation130,Citation131] and human cell atlas [Citation132] are illuminating receptor-specific expression profiles. One particularly exciting recent example is the chemokine receptor CCR8, which has restricted expression in Tregs that are in the tumor microenvironment [Citation108]. Therefore, with high-affinity antibodies, it is possible to deplete the immunosuppressive CCR8 positive Tregs from the tumor microenvironment specifically [Citation106]. This is an example of combining the unique expression profile of GPCRs with the properties of antibody therapeutics.
3.4. Extracellular G protein-coupled receptor interactions
Finally, because of their high affinity, binding via avidity and large size, antibody therapeutics are particularly suited to disrupt protein/protein interactions. Extracellular protein/protein interaction mapping (interactomics) has recently been highlighted for some of the extracellular proteins in the human genome [Citation133–136]. Checkpoint inhibitor therapies disrupt extracellular protein/protein interactions, and the extracellular interactions of GPCRs have only just begun to be mapped [Citation137]. Modulating extracellular interactions with antibodies would be informative to further understand the biology of adhesion GPCRs (also known as Class B2) and the various interactions involved between their extensive N terminal domains and other proteins. For example, the interaction between the adhesion GPCR called BAI1 and the leucine-rich repeat single transmembrane receptor called RTN4R was recently detailed to be critical for neuronal development [Citation138]. Since BAI1 is a tumor suppressor gene [Citation139], it may be possible to enhance or block this interaction with antibodies in order to activate BAI1 and slow tumor growth. The full capabilities of antibodies, such as inducing clustering or blocking extracellular interactions () is only just beginning to be explored for GPCRs.
4. Conclusion
Targeting GPCRs with antibodies and related biologics provides a unique opportunity for drug development. GPCRs are central to many physiological processes and are the target of a third of all FDA approved drugs, yet these drugs only target roughly 100 GPCRs. Biologics allow for GPCR de-orphanization, modulation of GPCR signaling, targeting of disease cells overexpressing GPCRs and disruption of GPCR interactions. However, only three clinically approved molecules have made it to the market. This is due to the inherent incompatibility of GPCRs with antibodies, namely, receptors have low surface expression and minimal exposed surface area. The development of GPCR biologics for clinical utility will enable new avenues for the treatment of many diseases.
5. Expert opinion
GPCRs are a challenging antigen for antibody binding because of their limited extracellular surface, conformational heterogeneity and cryptic-binding sites. That being said, we have reviewed some examples of autoantibodies [Citation20–22] that have been discovered to activate or inhibit GPCRs. Furthermore, antibodies have been developed and used in the clinic as drugs to treat diabetes, obesity, migraine, and cancer [Citation11,Citation54,Citation76]. In this review, we have described antibody drugs with traditional small-molecule pharmacology (agonism, antagonism, allosterism, etc.). However, many antibodies are profiled for binding before activation screening, and the best binding molecules typically bind to stable receptor conformations [Citation140]. Since agonism, inverse agonism, allosterism, and sometimes antagonism require stabilization of transition states, the binding of these complex pharmacophores is often poor, when compared to EC50 measured in functional assays [Citation141]. One interesting solution to this problem of highly efficacious molecules showing poor binding has been to engineer bivalent molecules, this was described above for ChemR23 [Citation67] and CXCR2 [Citation89] where a poor binding, but highly effective antagonist antibody fragment was paired with a strongly binding but poorly effective antagonist antibody fragment to create a highly potent antagonist biparatopic antibody with strong binding. It may be possible to engineer such bivalent or multivalent antibodies to tune the signaling of the receptor.
We have described many peptide fusions, where the antibody is targeted to a receptor or cell expressing the receptor of interest and the fused peptide is able to activate the receptor of interest, in particular, GLP-1 R for the treatment of diabetes and obesity [Citation11,Citation14–18]. This strategy is highly effective for peptide receptors where a rigid alpha helix is formed and the N-terminus of the peptide binds to the orthosteric site of the receptor, as is the case for GLP-1 R. However, another strategy would be to target the orthosteric site directly with the antibody CDRs, by using full-length receptors as antigens, as was demonstrated for APJR [Citation26]. Still another approach is the one used for ChemR23, where the extracellular loop 3 peptide was used as the antigen, and an agonist was found from screening the top-binding clones [Citation33]. The orthosteric site is particularly difficult to target with a CDR, because it is buried deep within the transmembrane bundles and the orthosteric site is where the energy of binding is transmitted to signal transducers such as G proteins. The orthosteric site is conformationally heterogeneous, so the CDRs must both fit into the orthosteric site and also stabilize a desired conformation (such as an agonist conformation).
The full utility of antibody engineering has only begun to be explored for GPCR-targeting antibodies. The bispecific molecules we described take advantage of tumor accumulation of antibodies, mixed with an immune system activating molecule. Using chimeric receptors to target engineered cells or viruses is a promising evolution of the idea behind bispecific molecules. We described BELMO, a chimeric adhesion GPCR that linked receptor binding to designer signaling pathways to treat inflammation. Previously, GPCR engineering strategies have proven utility in the so-called DREADDs [Citation142] and OptoXRs [Citation143]. In this way, it may be possible to engineer cell therapies that express GPCRs that are activated by blue light or an inert ligand. Combining engineered GPCRs with engineered cell therapies and antibodies that target GPCRs would add another tool to the available cell engineering toolkit.
Article highlights
The human genome contains over 800 G protein-coupled receptors (GPCRs).
GPCRs are central to many physiological processes and are the target of a third of all FDA approved drugs.
While GPCRs have already been targeted by many small molecules, antibodies and related biologics provide a unique opportunity for GPCR drug discovery.
Unlike small molecules, antibody therapeutics can easily be targeted to tissues, block extracellular protein interactions, and bind to multiple targets.
GPCRs remain difficult to target with antibodies despite antibody therapeutics offering unique drug properties.
The development of GPCR biologics for clinical utility will enable new avenues for the treatment of many diseases.
This box summarizes key points contained in the article.
Declaration of Interest
SM Peterson, AK Sato, CF Hu, M Mathur, JW Salameh and F Axelrod are all employees of Twist Bioscience. CJ Hutchings is an independent consultant who provides consultancy services to Abalone Bio, AbCellera Biologics, Abilita Bio, AdAlta, Confo Therapeutics, DJS Antibodies, Kyowa Kirin, OmniAb, Sosei-Heptares, Twist Biopharma and xCella Biosciences. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Dalesio NM, Barreto Ortiz SF, Pluznick JL, et al. Olfactory, taste, and photo sensory receptors in non-sensory organs: it just makes sense. Front Physiol. 2018;9:1673. PubMed PMID: 30542293; PubMed Central PMCID: PMCPMC6278613. eng.
- Munk C, Isberg V, Mordalski S, et al. GPCRdb: the G protein-coupled receptor database - an introduction. Br J Pharmacol. 2016 Jul;173(14):2195–2207. PubMed PMID: 27155948; PubMed Central PMCID: PMCPMC4919580. eng
- Einspahr JM, Tilley DG. Pathophysiological impact of the adhesion G protein-coupled receptor family. Am J Physiol Cell Physiol. 2022 Aug 1;323(2):C640–c647. PubMed PMID: 35848619; PubMed Central PMCID: PMCPMC9359651. eng
- Makurvet FD. Biologics vs. small molecules: drug costs and patient access. Med Drug Dis. 2021 March 01;9:100075.
- Brekke OH, Thommesen JE. Tailoring natural effector functions. Antibody engineering beyond humanization. Methods Mol Biol. 2003:207:383–391. PubMed PMID: 12412487; eng
- Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017 Sep;6(9):576–588. PubMed PMID: 28653357; PubMed Central PMCID: PMCPMC5613179. eng
- Stafford RL, Matsumoto ML, Yin G, et al. In vitro Fab display: a cell-free system for IgG discovery. Protein Eng Des Sel. 2014 Apr;27(4):97–109. PubMed PMID: 24586053; PubMed Central PMCID: PMCPMC3966677. eng
- Hutchings CJ. A review of antibody-based therapeutics targeting G protein-coupled receptors: an update. Expert Opin Biol Ther. 2020 Aug;20(8):925–935. PubMed PMID: 32264722; eng
- de Graaf C, Foata N, Engkvist O, et al. Molecular modeling of the second extracellular loop of G-protein coupled receptors and its implication on structure-based virtual screening. Proteins. 2008 May 1;71(2):599–620.
- Schädel SA, Heck M, Maretzki D, et al. Ligand channeling within a G-protein-coupled receptor. The entry and exit of retinals in native opsin. J Biol Chem. 2003 Jul 4;278(27):24896–24903.
- Glaesner W, Mark Vick A, Millican R, et al. Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an Fc fusion protein. Diabetes Metab Res Rev. 2010;26(4):287–296.
- Firan M, Bawdon R, Radu C, et al. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int Immunol. 2001 Aug;13(8):993–1002. PubMed PMID: 11470769; eng
- Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012 Dec;8(12):728–742. PubMed PMID: 22945360; eng
- Li C, Yang M, Wang X, et al. Glutazumab, a novel long-lasting GLP-1/anti-GLP-1R antibody fusion protein, exerts anti-diabetic effects through targeting dual receptor binding sites. Biochem Pharmacol. 2018 Apr;150: 46–53. PubMed PMID: 29355505; eng.
- Pan H, Su Y, Xie Y, et al. Everestmab, a novel long-acting GLP-1/anti GLP-1R nanobody fusion protein, exerts potent anti-diabetic effects. Artif Cells Nanomed Biotechnol. 2020 Dec;48(1):854–866. PubMed PMID: 32468873; eng.
- Liu Q, Garg P, Hasdemir B, et al. Functional GLP-1R antibodies identified from a synthetic GPCR-focused library demonstrate potent blood glucose control. MAbs. 2021 Jan-Dec;13(1):1893425. PubMed PMID: 33706686; PubMed Central PMCID: PMCPMC7971233. eng.
- Lu SC, Chen M, Atangan L, et al. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep Med. 2021 May 18;2(5):100263.
- Killion EA, Wang J, Yie J, et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med. 2018 Dec 19;10(472). PubMed PMID: 30567927; eng. doi: 10.1126/scitranslmed.aat3392.
- Cheloha RW, Fischer FA, Gardella TJ, et al. Activation of a G protein-coupled receptor through indirect antibody-mediated tethering of ligands. RSC Chem Biol. 2021 Dec 2;2(6):1692–1700. PubMed PMID: 34977584; PubMed Central PMCID: PMCPMC8637866. eng.
- Faust B, Billesbølle CB, Suomivuori CM, et al. Autoantibody mimicry of hormone action at the thyrotropin receptor. Nature. 2022 Sep;609(7928):846–853. PubMed PMID: 35940205; PubMed Central PMCID: PMCPMC9678024. eng.
- Chen CR, McLachlan SM, Hubbard PA, et al. Structure of a thyrotropin receptor monoclonal antibody variable region provides insight into potential mechanisms for its inverse agonist activity. Thyroid. 2018 Jul;28(7):933–940. PubMed PMID: 29845889; PubMed Central PMCID: PMCPMC6043401. eng
- Skiba MA, Kruse AC. Autoantibodies as Endogenous Modulators of GPCR Signaling. Trends Pharmacol Sci. 2021 Mar;42(3):135–150. PubMed PMID: 33358695; PubMed Central PMCID: PMCPMC7880908. eng
- Scholler P, Nevoltris D, de Bundel D, et al. Allosteric nanobodies uncover a role of hippocampal mGlu2 receptor homodimers in contextual fear consolidation. Nat Commun. 2017 Dec 6;8(1):1967.
- Meng J, Xu C, Lafon PA, et al. Nanobody-based sensors reveal a high proportion of mGlu heterodimers in the brain. Nat Chem Biol. 2022 Aug;18(8):894–903. PubMed PMID: 35681029; eng.
- Ullmer C, Zoffmann S, Bohrmann B, et al. Functional monoclonal antibody acts as a biased agonist by inducing internalization of metabotropic glutamate receptor 7. Br J Pharmacol. 2012 Dec;167(7):1448–1466. PubMed PMID: 22747985; PubMed Central PMCID: PMCPMC3514759. eng
- Ma Y, Ding Y, Song X, et al. Structure-guided discovery of a single-domain antibody agonist against human apelin receptor. Sci Adv. 2020 Jan;6(3):eaax7379. PubMed PMID: 31998837; PubMed Central PMCID: PMCPMC6962038. eng.
- Hutchings CJ, Cseke G, Osborne G, et al. Monoclonal anti-β1-adrenergic receptor antibodies activate G protein signaling in the absence of β-arrestin recruitment. MAbs. 2014 Jan-Feb;6(1):246–261. PubMed PMID: 24253107; PubMed Central PMCID: PMCPMC3929447. eng
- Jahns R, Boivin V, Lohse MJ. beta(1)-Adrenergic receptor function, autoimmunity, and pathogenesis of dilated cardiomyopathy. Trends Cardiovasc Med. 2006 Jan;16(1):20–24. PubMed PMID: 16387626; eng
- Jahns R, Boivin V, Krapf T, et al. Modulation of beta1-adrenoceptor activity by domain-specific antibodies and heart failure-associated autoantibodies. J Am Coll Cardiol. 2000 Oct;36(4):1280–1287. PubMed PMID: 11028484; eng
- Christ T, Schindelhauer S, Wettwer E, et al. Interaction between autoantibodies against the beta1-adrenoceptor and isoprenaline in enhancing L-type Ca2+ current in rat ventricular myocytes. J Mol Cell Cardiol. 2006 Oct;41(4):716–723. PubMed PMID: 16889792; eng
- Mobini R, Fu M, Wallukat G, et al. A monoclonal antibody directed against an autoimmune epitope on the human beta1-adrenergic receptor recognized in idiopathic dilated cardiomyopathy. Hybridoma. 2000 Apr;19(2):135–142. PubMed PMID: 10868793; eng
- Ishchenko A, Wacker D, Kapoor M, et al. Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc Natl Acad Sci U S A. 2017 Aug 1;114(31):8223–8228.
- Trilleaud C, Gauttier V, Biteau K, et al. Agonist anti-ChemR23 mAb reduces tissue neutrophil accumulation and triggers chronic inflammation resolution. Sci Adv. 2021 Apr;7(14). PubMed PMID: 33811066; eng. doi: 10.1126/sciadv.abd1453.
- Kamel R, Eftekhari P, Garcia S, et al. A high-affinity monoclonal antibody with functional activity against the 5-hydroxytryptaminergic (5-HT4) receptor. Biochem Pharmacol. 2005 Oct 1;70(7):1009–1018.
- Gupta A, Décaillot FM, Gomes I, et al. Conformation state-sensitive antibodies to G-protein-coupled receptors. J Biol Chem. 2007 Feb 23;282(8):5116–5124.
- Gomes I, Gupta A, Singh SP, et al. Monoclonal antibody to the delta opioid receptor acts as an agonist in dual regulation of adenylate cyclase in NG108-15 cells. FEBS Lett. 1999 Jul 30;456(1):126–130.
- Lebesgue D, Wallukat G, Mijares A, et al. An agonist-like monoclonal antibody against the human beta2-adrenoceptor. Eur J Pharmacol. 1998 May 1;348(1):123–133.
- Mijares A, Lebesgue D, Wallukat G, et al. From agonist to antagonist: fab fragments of an agonist-like monoclonal anti-beta(2)-adrenoceptor antibody behave as antagonists. Mol Pharmacol. 2000 Aug;58(2):373–379. PubMed PMID: 10908305; eng
- Schreiber AB, Couraud PO, Andre C, et al. Anti-alprenolol anti-idiotypic antibodies bind to beta-adrenergic receptors and modulate catecholamine-sensitive adenylate cyclase. Proc Natl Acad Sci U S A. 1980 Dec;77(12):7385–7389. PubMed PMID: 6261254; PubMed Central PMCID: PMCPMC350508. eng
- Guillet JG, Kaveri SV, Durieu O, et al. beta-Adrenergic agonist activity of a monoclonal anti-idiotypic antibody. Proc Natl Acad Sci U S A. 1985 Mar;82(6):1781–1784. PubMed PMID: 2858851; PubMed Central PMCID: PMCPMC397356. eng
- Elies R, Fu LX, Eftekhari P, et al. Immunochemical and functional characterization of an agonist-like monoclonal antibody against the M2 acetylcholine receptor. Eur J Biochem. 1998 Feb 1;251(3):659–666.
- Abu Alla S, Quitterer U, Grigoriev S, et al. Extracellular domains of the bradykinin B2 receptor involved in ligand binding and agonist sensing defined by anti-peptide antibodies. J Biol Chem. 1996 Jan 19;271(3):1748–1755.
- Goetzl EJ, Dembrow D, Van Brocklyn JR, et al. An IgM-kappa rat monoclonal antibody specific for the type 1 sphingosine 1-phosphate G protein-coupled receptor with antagonist and agonist activities. Immunol Lett. 2004 Apr 30;93(1):63–69.
- Wang CJ, Tai HH. Monoclonal anti-idiotypic antibodies to platelet activating factor (PAF) and their interaction with PAF receptors. J Biol Chem. 1991 Jul 5;266(19):12372–12378. PubMed PMID: 1648095; eng
- Blanpain C, Vanderwinden JM, Cihak J, et al. Multiple active states and oligomerization of CCR5 revealed by functional properties of monoclonal antibodies. Mol Biol Cell. 2002 Feb;13(2):723–737. PubMed PMID: 11854425; PubMed Central PMCID: PMCPMC65662. eng
- Hennen S, Kodra JT, Soroka V, et al. Structural insight into antibody-mediated antagonism of the Glucagon-like peptide-1 Receptor. Sci Rep. 2016 May 19;6:26236. PubMed PMID: 27196125; PubMed Central PMCID: PMCPMC4872540. eng.
- Ravn P, Madhurantakam C, Kunze S, et al. Structural and pharmacological characterization of novel potent and selective monoclonal antibody antagonists of glucose-dependent insulinotropic polypeptide receptor. J Biol Chem. 2013 Jul 5;288(27):19760–19772.
- Okamoto H, Kim J, Aglione J, et al. Glucagon receptor blockade with a human antibody normalizes blood glucose in diabetic mice and monkeys. Endocrinology. 2015 Aug;156(8):2781–2794. PubMed PMID: 26020795; eng.
- Kostic A, King TA, Yang F, et al. A first-in-human pharmacodynamic and pharmacokinetic study of a fully human anti-glucagon receptor monoclonal antibody in normal healthy volunteers. Diabetes Obes Metab. 2018 Feb;20(2):283–291. PubMed PMID: 28755409; PubMed Central PMCID: PMCPMC5813272. eng
- Murphy AJ, Macdonald LE, Stevens S, et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci U S A. 2014 Apr 8;111(14):5153–5158.
- Pettus J, Reeds D, Cavaiola TS, et al. Effect of a glucagon receptor antibody (REMD-477) in type 1 diabetes: a randomized controlled trial. Diabetes Obes Metab. 2018 May;20(5):1302–1305. PubMed PMID: 29283470; PubMed Central PMCID: PMCPMC6181222. eng.
- Pettus J, Boeder SC, Christiansen MP, et al. Glucagon receptor antagonist volagidemab in type 1 diabetes: a 12-week, randomized, double-blind, phase 2 trial. Nat Med. 2022 Oct;28(10):2092–2099. PubMed PMID: 36192552; eng
- van der Woning B, De Boeck G, Blanchetot C, et al. DNA immunization combined with scFv phage display identifies antagonistic GCGR specific antibodies and reveals new epitopes on the small extracellular loops. mAbs. 2016;8(6):1126–1135.
- Shi L, Lehto SG, Zhu DX, et al. Pharmacologic Characterization of AMG 334, a Potent and Selective Human Monoclonal Antibody against the Calcitonin Gene-Related Peptide Receptor. J Pharmacol Exp Ther. 2016 Jan;356(1):223–231. PubMed PMID: 26559125; eng.
- King CT, Gegg CV, Hu SN-Y, et al. Discovery of the Migraine Prevention Therapeutic Aimovig (Erenumab), the first FDA-approved antibody against a G-protein-coupled receptor. ACS Pharmacol Transl Sci. 2019;2(6):485–490.
- Garces F, Mohr C, Zhang L, et al. Molecular insight into recognition of the CGRPR complex by migraine prevention therapy aimovig (erenumab). Cell Rep. 2020;30(6):1714–1723.e6.
- Fischer MM, Cancilla B, Yeung VP, et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci Adv. 2017 Jun;3(6):e1700090. PubMed PMID: 28691093; PubMed Central PMCID: PMCPMC5479655. eng.
- Sasaki Y, Kosaka H, Usami K, et al. Establishment of a novel monoclonal antibody against LGR5. Biochem Biophys Res Commun. 2010 Apr 9;394(3):498–502.
- Zhang C, Wang X, Zhang H, et al. Therapeutic monoclonal antibody antagonizing endothelin receptor A for pulmonary arterial hypertension. J Pharmacol Exp Ther. 2019 Jul;370(1):54–61. PubMed PMID: 30992315; eng.
- Ju MS, Ahn HM, Han SG, et al. A human antibody against human endothelin receptor type A that exhibits antitumor potency. Exp Mol Med. 2021 Sep;53(9):1437–1448. PubMed PMID: 34588605; PubMed Central PMCID: PMCPMC8492878. eng.
- Douthwaite JA, Sridharan S, Huntington C, et al. Affinity maturation of a novel antagonistic human monoclonal antibody with a long VH CDR3 targeting the Class A GPCR formyl-peptide receptor 1. MAbs. 2015;7(1):152–166. PubMed PMID: 25484051; PubMed Central PMCID: PMCPMC4622665. eng.
- McMahon C, Staus DP, Wingler LM, et al. Synthetic nanobodies as angiotensin receptor blockers. Proc Natl Acad Sci U S A. 2020 Aug 18;117(33):20284–20291.
- Wellner A, McMahon C, Gilman MSA, et al. Rapid generation of potent antibodies by autonomous hypermutation in yeast. Nat Chem Biol. 2021 Oct;17(10):1057–1064. PubMed PMID: 34168368; PubMed Central PMCID: PMCPMC8463502. eng
- Westlund KN, Montera MA, Goins AE, et al. Single-chain Fragment variable antibody targeting cholecystokinin-B receptor for pain reduction. Neurobiol Pain. 2021 Aug-Dec;10: 100067. PubMed PMID: 34458647; PubMed Central PMCID: PMCPMC8378781. eng.
- Peter JC, Nicholson JR, Heydet D, et al. <atl>Antibodies. Am J Physiol Regul Integr Comp Physiol. 2007 Jun;292(6):R2151–8. PubMed PMID: 17322114; eng
- Soave M, Cseke G, Hutchings CJ, et al. A monoclonal antibody raised against a thermo-stabilised β(1)-adrenoceptor interacts with extracellular loop 2 and acts as a negative allosteric modulator of a sub-set of β(1)-adrenoceptors expressed in stable cell lines. Biochem Pharmacol. 2018 Jan;147:38–54.
- Peyrassol X, Laeremans T, Gouwy M, et al. Development by genetic immunization of monovalent antibodies (nanobodies) behaving as antagonists of the human ChemR23 receptor. J Immunol. 2016 Mar 15;196(6):2893–2901.
- Buchner RR, Vogen SM, Fischer W, et al. Anti-human kappa opioid receptor antibodies: characterization of site-directed neutralizing antibodies specific for a peptide kappa R(33-52) derived from the predicted amino terminal region of the human kappa receptor. J Immunol. 1997 Feb 15;158(4):1670–1680. PubMed PMID: 9029103; eng
- Gaich G, BILIC S, DAGON Y, et al. 392-P: phase 1, randomized, controlled trial of GFB-024, a once-monthly CB1 inverse agonist, in healthy overweight and obese participants and in participants with type 2 diabetes mellitus. Diabetes. 2022;71(Supplement_1). DOI:10.2337/db22-392-P
- Bilic S, Dagon Y, Gustafson T, et al. OR03-2 phase 1, randomized, controlled trial of GFB-024, a once-monthly CB1 inverse agonist, in healthy overweight and obese participants and in participants with type 2 diabetes mellitus. J Endocr Soc. 2022 Nov 1;6(Suppl 1):A348.
- Carvelli J, Meziani F, Dellamonica J, et al. Avdoralimab (Anti-C5aR1 mAb) versus placebo in patients with severe COVID-19: results from a randomized controlled trial (FOR COVID elimination [FORCE]). Crit Care Med. 2022 Dec 1;50(12):1788–1798.
- van der Pals J, Koul S, Andersson P, et al. Treatment with the C5a receptor antagonist ADC-1004 reduces myocardial infarction in a porcine ischemia-reperfusion model. BMC Cardiovasc Disord. 2010 Sep 27;10:45.
- Huang L, Sato AK, Sachdeva M, et al. Discovery of human antibodies against the C5aR target using phage display technology. J Mol Recognit. 2005 Jul-Aug;18(4):327–333. PubMed PMID: 15706605; eng
- Oppermann M, Raedt U, Hebell T, et al. Probing the human receptor for C5a anaphylatoxin with site-directed antibodies. Identification of a potential ligand binding site on the NH2-terminal domain. J Immunol. 1993 Oct 1;151(7):3785–3794. PubMed PMID: 8376805; eng
- Morgan EL, Ember JA, Sanderson SD, et al. Anti-C5a receptor antibodies. Characterization of neutralizing antibodies specific for a peptide, C5aR-(9-29), derived from the predicted amino-terminal sequence of the human C5a receptor. J Immunol. 1993 Jul 1;151(1):377–388. PubMed PMID: 8326131; eng
- Subramaniam JM, Whiteside G, McKeage K, et al. Mogamulizumab: first global approval. Drugs. 2012 Jun 18;72(9):1293–1298.
- Hagemann UB, Gunnarsson L, Géraudie S, et al. Fully human antagonistic antibodies against CCR4 potently inhibit cell signaling and chemotaxis. PLoS One. 2014;9(7):e103776.
- Chang DK, Sui J, Geng S, et al. Humanization of an anti-CCR4 antibody that kills cutaneous T-cell lymphoma cells and abrogates suppression by T-regulatory cells. Mol Cancer Ther. 2012 Nov;11(11):2451–2461. PubMed PMID: 22869555; PubMed Central PMCID: PMCPMC3496034. eng
- Han T, Abdel-Motal UM, Chang DK, et al. Human anti-CCR4 minibody gene transfer for the treatment of cutaneous T-cell lymphoma. PLoS One. 2012;7(9):e44455.
- Peng SB, Zhang X, Paul D, et al. Inhibition of CXCR4 by LY2624587, a fully humanized anti-CXCR4 antibody induces apoptosis of hematologic malignancies. PLoS One. 2016;11(3):e0150585. PubMed PMID: 26954567; PubMed Central PMCID: PMCPMC4782998. eng.
- Peng SB, Van Horn RD, Yin T, et al. Distinct mobilization of leukocytes and hematopoietic stem cells by CXCR4 peptide antagonist LY2510924 and monoclonal antibody LY2624587. Oncotarget. 2017 Nov 7;8(55):94619–94634.
- Kuhne MR, Mulvey T, Belanger B, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013 Jan 15;19(2):357–366.
- Griffiths K, Dolezal O, Cao B, et al. i-bodies, human single domain antibodies that antagonize chemokine receptor CXCR4. J Biol Chem. 2016 Jun 10;291(24):12641–12657.
- Peng L, Damschroder MM, Cook KE, et al. Molecular basis for the antagonistic activity of an anti-CXCR4 antibody. MAbs. 2016;8(1):163–175.
- Jähnichen S, Blanchetot C, Maussang D, et al. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc Natl Acad Sci U S A. 2010 Nov 23;107(47):20565–20570.
- Soave M, Heukers R, Kellam B, et al. Monitoring Allosteric Interactions with CXCR4 Using NanoBiT Conjugated Nanobodies. Cell Chem Biol. 2020 Oct 15;27(10):1250–1261.e5.
- Endres MJ, Clapham PR, Marsh M, et al. CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell. 1996 Nov 15;87(4):745–756.
- Maussang D, Mujić-Delić A, Descamps FJ, et al. Llama-derived single variable domains (nanobodies) directed against chemokine receptor CXCR7 reduce head and neck cancer cell growth in vivo. J Biol Chem. 2013 Oct 11;288(41):29562–29572.
- Bradley ME, Dombrecht B, Manini J, et al. Potent and efficacious inhibition of CXCR2 signaling by biparatopic nanobodies combining two distinct modes of action. Mol Pharmacol. 2015 Feb;87(2):251–262. PubMed PMID: 25468882; eng
- Rossant CJ, Carroll D, Huang L, et al. Phage display and hybridoma generation of antibodies to human CXCR2 yields antibodies with distinct mechanisms and epitopes. MAbs. 2014;6(6):1425–1438.
- Osbourn JK, Earnshaw JC, Johnson KS, et al. Directed selection of MIP-1 alpha neutralizing CCR5 antibodies from a phage display human antibody library. Nat Biotechnol. 1998 Aug;16(8):778–781. PubMed PMID: 9702779; eng
- Mirzabekov T, Kontos H, Farzan M, et al. Paramagnetic proteoliposomes containing a pure, native, and oriented seven-transmembrane segment protein, CCR5. Nat Biotechnol. 2000 Jun;18(6):649–654. PubMed PMID: 10835604; eng
- Sommerfelt MA. Circular CCR5 peptide conjugates and uses thereof (WO2008074895). Expert Opin Ther Pat. 2009 Sep;19(9):1323–1328. PubMed PMID: 19456281; eng
- Lalezari J, Yadavalli GK, Para M, et al. Safety, pharmacokinetics, and antiviral activity of HGS004, a novel fully human IgG4 monoclonal antibody against CCR5, in HIV-1-infected patients. J Infect Dis. 2008 Mar 1;197(5):721–727.
- Khatib N, Das S. PRO 140–a novel CCR5 co-receptor inhibitor. Recent Pat Antiinfect Drug Discov. 2010 Jan;5(1):18–22. PubMed PMID: 19807678; eng
- Vergunst CE, Gerlag DM, Lopatinskaya L, et al. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2008 Jul;58(7):1931–1939. PubMed PMID: 18576354; eng
- Gilbert J, Lekstrom-Himes J, Donaldson D, et al. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am J Cardiol. 2011 Mar 15;107(6):906–911.
- Somovilla-Crespo B, Alfonso-Pérez M, Cuesta-Mateos C, et al. Anti-CCR7 therapy exerts a potent anti-tumor activity in a xenograft model of human mantle cell lymphoma. J Hematol Oncol. 2013 Dec 4;6:89.
- Chamorro S, Vela M, Franco-Villanueva A, et al. Antitumor effects of a monoclonal antibody to human CCR9 in leukemia cell xenografts. MAbs. 2014 Jul-Aug;6(4):1000–1012. PubMed PMID: 24870448; PubMed Central PMCID: PMCPMC4171004. eng
- Heukers R, Fan TS, de Wit RH, et al. The constitutive activity of the virally encoded chemokine receptor US28 accelerates glioblastoma growth. Oncogene. 2018 Jul;37(30):4110–4121. PubMed PMID: 29706656; PubMed Central PMCID: PMCPMC6062493. eng
- De Groof TWM, Mashayekhi V, Fan TS, et al. Nanobody-targeted photodynamic therapy selectively kills viral GPCR-expressing glioblastoma cells. Mol Pharm. 2019 Jul 1;16(7):3145–3156.
- Carter PJ, Rajpal A. Designing antibodies as therapeutics. Cell. 2022 Jul 21;185(15):2789–2805. PubMed PMID: 35868279; eng
- Ishii T, Ishida T, Utsunomiya A, et al. Defucosylated humanized anti-CCR4 monoclonal antibody KW-0761 as a novel immunotherapeutic agent for adult T-cell leukemia/lymphoma. Clin Cancer Res. 2010 Mar 1;16(5):1520–1531.
- Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs. 2012 Jul-Aug;4(4):3145–3156. PubMed PMID: 22699226; PubMed Central PMCID: PMCPMC3499336. eng
- Van Damme H, Dombrecht B, Kiss M, et al. Therapeutic depletion of CCR8(+) tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J Immunother Cancer. 2021 Feb;9(2). PubMed PMID: 33589525; PubMed Central PMCID: PMCPMC7887378. eng. doi: 10.1136/jitc-2020-001749.
- Weaver JD, Stack EC, Buggé JA, et al. Differential expression of CCR8 in tumors versus normal tissue allows specific depletion of tumor-infiltrating T regulatory cells by GS-1811, a novel Fc-optimized anti-CCR8 antibody. Oncoimmunology. 2022;11(1):2141007. PubMed PMID: 36352891; PubMed Central PMCID: PMCPMC9639568. eng.
- Campbell JR, McDonald BR, Mesko PB, et al. Fc-optimized anti-CCR8 antibody depletes regulatory T cells in human tumor models. Cancer Res. 2021 Jun 1;81(11):2983–2994.
- Whiteside SK, Grant FM, Gyori DS, et al. CCR8 marks highly suppressive Treg cells within tumours but is dispensable for their accumulation and suppressive function. Immunology. 2021 Aug;163(4):512–520. PubMed PMID: 33838058; PubMed Central PMCID: PMCPMC8274197. eng
- Herpers B, Eppink B, James MI, et al. Functional patient-derived organoid screenings identify MCLA-158 as a therapeutic EGFR × LGR5 bispecific antibody with efficacy in epithelial tumors. Nat Cancer. 2022 Apr;3(4):418–436. PubMed PMID: 35469014; eng.
- Hao S, Xu S, Li L, et al. Tumour inhibitory activity on pancreatic cancer by bispecific nanobody targeting PD-L1 and CXCR4. BMC Cancer. 2022 Oct 25;22(1):1092.
- Verkleij CPM, Broekmans MEC, van Duin M, et al. Preclinical activity and determinants of response of the GPRC5DxCD3 bispecific antibody talquetamab in multiple myeloma. Blood Adv. 2021 Apr 27;5(8):2196–2215.
- Panjideh H, Müller G, Koch M, et al. Immunotherapy of B-cell non-Hodgkin lymphoma by targeting the chemokine receptor CXCR5 in a preclinical mouse model. Int J Cancer. 2014 Dec 1;135(11):2623–2632.
- Kularatne SA, Deshmukh V, Ma J, et al. A CXCR4-targeted site-specific antibody-drug conjugate. Angew Chem Int Ed Engl. 2014 Oct 27;53(44):11863–11867.
- Costa MJ, Kudaravalli J, Ma JT, et al. Optimal design, anti-tumour efficacy and tolerability of anti-CXCR4 antibody drug conjugates. Sci Rep. 2019 Feb 21;9(1):2443.
- Wang RE, Liu T, Wang Y, et al. An immunosuppressive antibody-drug conjugate. J Am Chem Soc. 2015 Mar 11;137(9):3229–3232.
- Cunha-Santos C, Perdigao PRL, Martin F, et al. Inhibition of HIV replication through siRNA carried by CXCR4-targeted chimeric nanobody. Cell Mol Life Sci. 2020 Jul;77(14):2859–2870. PubMed PMID: 31641784; eng.
- Mailankody S, Devlin SM, Landa J, et al. GPRC5D-Targeted CAR T Cells for Myeloma. N Engl J Med. 2022 Sep 29;387(13):1196–1206.
- Sanchez Gil J, Dubois M, Neirinckx V, et al. Nanobody-based retargeting of an oncolytic herpesvirus for eliminating CXCR4(+) GBM cells: a proof of principle. Mol Ther Oncolytics. 2022 Sep 15;26:35–48. PubMed PMID: 35784400; PubMed Central PMCID: PMCPMC9217993. eng.
- Morioka S, Kajioka D, Yamaoka Y, et al. Chimeric efferocytic receptors improve apoptotic cell clearance and alleviate inflammation. Cell. 2022 Dec 22;185(26):4887–4903.e17.
- Hauser AS, Gloriam DE, Brauner-Osborne H, et al. Novel approaches leading towards peptide GPCR de-orphanisation. Br J Pharmacol. 2020 Mar;177(5):961–968. PubMed PMID: 31863461; PubMed Central PMCID: PMCPMC7042120
- Olsen RHJ, DiBerto JF, English JG, et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat Chem Biol. 2020 Aug;16(8):841–849. PubMed PMID: 32367019; PubMed Central PMCID: PMCPMC7648517. eng
- Paek J, Kalocsay M, Staus DP, et al. Multidimensional tracking of GPCR signaling via peroxidase-catalyzed proximity labeling. Cell. 2017 Apr 6;169(2):338–349.e11.
- Lobingier BT, Hüttenhain R, Eichel K, et al. An approach to spatiotemporally resolve protein interaction networks in living cells. Cell. 2017 Apr 6;169(2):350–360.e12.
- Kroeze WK, Sassano MF, Huang XP, et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol. 2015 May;22(5):362–369. PubMed PMID: 25895059; PubMed Central PMCID: PMCPMC4424118. eng
- Watkins LR, Orlandi C. In vitro profiling of orphan G protein coupled receptor (GPCR) constitutive activity. Br J Pharmacol. 2021 Aug;178(15):2963–2975. PubMed PMID: 33784795; eng
- Wingler LM, Lefkowitz RJ. Conformational basis of G protein-coupled receptor signaling versatility. Trends Cell Biol. 2020 Sep;30(9):736–747. PubMed PMID: 32622699; PubMed Central PMCID: PMCPMC7483927. eng
- Webb DR, Handel TM, Kretz-Rommel A, et al. Opportunities for functional selectivity in GPCR antibodies. Biochem Pharmacol. 2013 Jan 15;85(2):147–152.
- Yuan TZ, Garg P, Wang L, et al. Rapid discovery of diverse neutralizing SARS-CoV-2 antibodies from large-scale synthetic phage libraries. MAbs. 2022 Jan-Dec;14(1):2002236. PubMed PMID: 34967699; PubMed Central PMCID: PMCPMC8726723. eng
- Laeremans T, Sands ZA, Claes P, et al. Accelerating GPCR drug discovery with conformation-stabilizing VHHs. Front Mol Biosci. 2022;9:863099. PubMed PMID: 35677880; PubMed Central PMCID: PMCPMC9170359. eng
- Karlsson M, Zhang C, Méar L, et al. A single-cell type transcriptomics map of human tissues. Sci Adv. 2021 Jul;7(31). doi: 10.1126/sciadv.abh2169. PubMed PMID: 34321199; PubMed Central PMCID: PMCPMC8318366. eng
- Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015 Jan 23;347(6220):1260419.
- Rood JE, Maartens A, Hupalowska A, et al. Impact of the human cell atlas on medicine. Nat Med. 2022 Dec 8. PubMed PMID: 36482102; eng. doi: 10.1038/s41591-022-02104-7
- Verschueren E, Husain B, Yuen K, et al. The immunoglobulin superfamily receptome defines cancer-relevant networks associated with clinical outcome. Cell. 2020 Jul 23;182(2):329–344.e19.
- Wojtowicz WM, Vielmetter J, Fernandes RA, et al. A human IgSF cell-surface interactome reveals a complex network of protein-protein interactions. Cell. 2020 Aug 20;182(4):1027–1043.e17.
- Siepe DH, Henneberg LT, Wilson SC, et al. Identification of orphan ligand-receptor relationships using a cell-based CRISPRa enrichment screening platform. Elife. 2022 Sep 30;11. PubMed PMID: 36178190; PubMed Central PMCID: PMCPMC9578707. eng. 10.7554/eLife.81398
- Chong ZS, Ohnishi S, Yusa K, et al. Pooled extracellular receptor-ligand interaction screening using CRISPR activation. Genome Biol. 2018 Nov 26;19(1):205.
- Dunn HA, Orlandi C, Martemyanov KA. Beyond the ligand: extracellular and transcellular G protein-coupled receptor complexes in physiology and pharmacology. Pharmacol Rev. 2019 Oct;71(4):503–519. PubMed PMID: 31515243; PubMed Central PMCID: PMCPMC6742926. eng.
- Wang J, Miao Y, Wicklein R, et al. RTN4/NoGo-receptor binding to BAI adhesion-GPCRs regulates neuronal development. Cell. 2021 Nov 24;184(24):5869–5885.e25.
- Zhu D, Osuka S, Zhang Z, et al. BAI1 suppresses medulloblastoma formation by protecting p53 from Mdm2-mediated degradation. Cancer Cell. 2018 Jun 11;33(6):1004–1016.e5.
- Weis WI, Kobilka BK. The molecular basis of G protein-coupled receptor activation. Annu Rev Biochem. 2018 Jun 20; 87:897–919. PubMed PMID: 29925258; PubMed Central PMCID: PMCPMC6535337. eng
- Kenakin TP. The quantification of relative efficacy of agonists. J Pharmacol Methods. 1985;13(4):281–308.
- Roth BL. DREADDs for neuroscientists. Neuron. 2016 Feb 17;89(4):683–694. PubMed PMID: 26889809; PubMed Central PMCID: PMCPMC4759656. eng
- Tichy AM, Gerrard EJ, Sexton PM, et al. Light-activated chimeric GPCRs: limitations and opportunities. Curr Opin Struct Biol. 2019 Aug;57:196–203.