
ABSTRACT
Introduction: Nowadays, one of the most serious treatment complications in hemophilia A is the formation of neutralizing antibodies against coagulation factor VIII (FVIII). These so-called inhibitors develop in about 30% of all patients with severe hemophilia A. Once formed, inhibitors reduce FVIII efficacy in blood coagulation, which has a negative impact on patients’ health and quality of life and significantly increases hemophilia A treatment costs. The pathophysiology of inhibitor development is a complex and multi-causal process, in which both genetic factors as well as environmental factors participate. So-called ‘danger signals’ are considered contributors to inhibitor formation, and can be triggered by surgery, joint bleeds or infections. A pro-inflammatory tissue micro-environment is thereby established, which is characterized by the upregulation of costimulatory molecules on antigen-presenting cells (APCs), that can facilitate the alloimmunization to FVIII and thereby inhibitor formation. Here, the authors will discuss evidence from (pre)clinical studies about this theory in hemophilia A.
Areas covered: In this review, the current knowledge regarding the ‘danger theory’ with regard to inhibitor development in hemophilia A is summarized.
Expert opinion: Danger signals might contribute to inhibitor development; however, the evidence is scarce and not conclusive. Future studies, like multinational registries, are warranted but challenging.
1. Introduction
Hemophilia A is a severe X-linked congenital bleeding disorder, caused by a deficit in the coagulation protein factor VIII (FVIII) and affects approximately 1 in 5000 males worldwide [Citation1]. Due to the lack of FVIII, patients suffer from prolonged and serious, sometimes life-threatening bleeding, and require frequent FVIII replacement therapy in order to prevent or treat these bleeding events. Nowadays the most significant complication of hemophilia treatment is the development of neutralizing antibodies against FVIII [Citation2]. These so-called ‘inhibitors’ occur in approximately 30% of all patients with severe hemophilia A [Citation3,Citation4]. As a result of these inhibitors, traditional replacement therapy becomes ineffective, necessitating the use of bypassing agents. This subsequently leads to a significant increase in morbidity and treatment costs, and negatively influences patients’ quality of life [Citation5,Citation6].
Eradication of the anti-FVIII antibodies is possible by Immune Tolerance Induction (ITI), whereby repeated and long-term administration of FVIII ultimately results in a downregulation of the neutralizing antibody production in about 70% of all cases [Citation7–Citation9]. However, ITI is an invasive and costly therapy. Much effort focuses on the prevention of inhibitor development. Therefore, understanding the pathophysiology of inhibitor development is needed.
The intriguing question why some patients do develop inhibitors and others do not, is still unresolved. Both genetic and environmental factors appear to be involved. While the correlative relationship between the severity of the F8 gene mutation and inhibitor risk is well established, this is less clear for most environmental risk factors. Theoretically, an interesting environmental risk factor is the presence of ‘danger signals’, like surgery or infections, during FVIII administration. According to the ‘danger theory’ these immune system alert signals result in the upregulation of costimulatory molecules on antigen-presenting cells (APCs), enabling them to fully activate naïve CD4 T-cells, which provide help to antibody-producing B-cells and ultimately lead to the production of anti-FVIII antibodies [Citation10]. Contrary to this theoretical base, the evidence regarding danger signals as potential risk factors for inhibitor development is scarce. This review aims at providing an update on the current knowledge about the role of danger signals in inhibitor development in hemophilia A. Firstly, the pathophysiology of alloimmunization to FVIII and theoretical concepts of the danger theory are described. Thereafter, data from preclinical and clinical studies are summarized to discuss to what extent the danger theory applies to inhibitor development in hemophilia A.
2. Pathophysiology of inhibitor development
2.1. Immune response to FVIII
The pathogenesis of FVIII inhibitor formation is a complex and multi-causal process, involving the interaction of both genetic and environmental factors. Allo-immunization to FVIII requires the presentation of FVIII peptides in complex with molecules of the major histocompatibility complex (MHC) by antigen presenting cells (APCs) to T-cells, and the subsequent activation of FVIII-specific B-cells, ultimately resulting in the production of anti-FVIII antibodies () [Citation11–Citation15]. Due to the reduced level of FVIII or abnormalities in its configuration in hemophilia A, central and peripheral immune tolerance to this coagulation protein is not as established as in healthy individuals. Consequently, this could lead to the presence of FVIII-specific B- and T-cells and the risk of activating these cells upon FVIII administration.
Figure 1. Immune response to FVIII based on the danger theory.
I. Factor VIII (FVIII) administration in absence of danger signals: After administration, FVIII is internalized by antigen presenting cells (APCs), e.g. dendritic cells (DCs), and is presented to naïve CD4 + T-cells. In absence of danger signals and thus no stimulation of the pattern recognition receptors (PRR), co-stimulatory molecules on the DC are not upregulated. This results in a non-productive immunologic synapse between the DC and the T-cell, whereby T-cells become anergic and are not able to stimulate B-cells.II. FVIII administration in the presence of danger signals: Several events, for example, joint bleeds, can result in tissue damage and the subsequent release of damage-associated molecular patterns (DAMPs). These DAMPS can bind to PRR on the DC, resulting in the upregulation of costimulatory molecules (CD80/CD86) and adhesion molecules, and the release of immune stimulatory cytokines. Together with the presentation of FVIII on the Major Histocompatibility (MHC) class II complex, these signals activate naïve CD4 + T-cells. In turn, these activated T-cells activate FVIII-specific naive B-cells, which expand and differentiate either into plasma cells, secreting anti-FVIII antibodies (FVIII plasma cell), or FVIII-specific B-memory cells (FVIII B-mem). FVIII-specific T-memory (T mem) cells are also formed.CD40L: CD40 ligand; TCR: T-cell receptor. BCR: B-cell receptor.
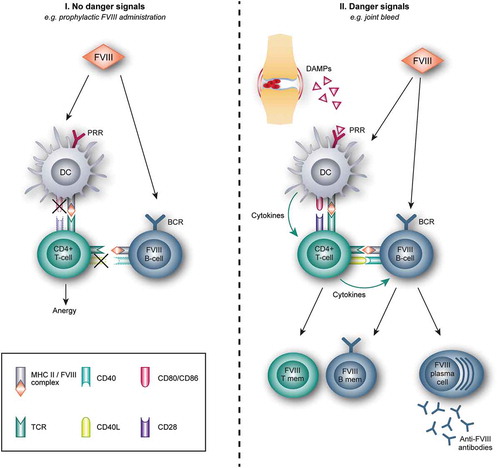
In more detail, the process of inhibitor formation starts with APCs, mostly dendritic cells (DCs), which endocytose the encountered FVIII. Although the exact mechanisms of endocytosis still need to be clarified, both in vitro as in vivo studies indicate that FVIII enters APCs by its C1 domain and that the macrophage mannose receptor (MMR) on DCs in involved in this process [Citation16–Citation18]. After internalization, FVIII is processed into peptides and presented on the MHC class II molecules to naïve CD4 T-cells in secondary lymphoid organs [Citation19,Citation20]. In addition to FVIII-derived peptide/MHC complex presentation, co-stimulatory signals are necessary to fully activate naïve T-cells able to bind FVIII epitopes. A pro-inflammatory micro-environment will provide this required co-stimulation. The presence of danger-/damage- and pathogen-associated molecular patterns (DAMPS and PAMPs, respectively) results in the upregulation of co-stimulatory molecules, most notably the CD80 and CD86 molecules, adhesion molecules and the release of pro-inflammatory cytokines.
Activated naïve FVIII-specific T-cells consequently differentiate into effector helper CD4 + T-cells, which in turn activate FVIII-specific B-cells, allowing them to undergo class switching and differentiate into FVIII-specific memory B-cells and anti-FVIII producing plasma cells.
So far, the hypothesized pathogenesis of FVIII inhibitor development mainly follows the classical immune response to ‘foreign’ components. However, one important and still unresolved question remains: Why do some patients and others not develop inhibitors?
2.2. Lowering the threshold of tolerance to FVIII treatment
In an attempt to answer this question many studies have been performed, resulting in the identification of several risk factors, which together cover the amount and configuration of the FVIII protein itself, its interaction with the immune system and the inflammatory state during FVIII administration () [Citation4,Citation21–Citation23].
Figure 2. Risk factors of inhibitor formation in hemophilia A [Citation3,Citation4,Citation22,Citation24,Citation26–Citation29,Citation34–Citation37,Citation73,Citation76,Citation78,Citation79,Citation94–Citation100].
Indicative overview of genetic and environmental risk factors in relation to the level of evidence. *The role of FVIII product type and the risk of inhibitor development is a highly debated issue, whereby recent studies are unequivocal.
![Figure 2. Risk factors of inhibitor formation in hemophilia A [Citation3,Citation4,Citation22,Citation24,Citation26–Citation29,Citation34–Citation37,Citation73,Citation76,Citation78,Citation79,Citation94–Citation100].Indicative overview of genetic and environmental risk factors in relation to the level of evidence. *The role of FVIII product type and the risk of inhibitor development is a highly debated issue, whereby recent studies are unequivocal.](/cms/asset/ddaded48-d536-49ae-b100-7bac66b3a55a/ierr_a_1604213_f0002_oc.jpg)
These risk factors can be divided into genetic factors, like the F8 gene mutation and polymorphisms in immune response genes, and environmental factors, including the FVIII product type and treatment regimen, the intensity of treatment at first exposure and the presence of danger signals during administration [Citation4,Citation22–Citation26].
So far the best predictive factor for inhibitor development is the severity of hemophilia A and the F8 gene mutation. Hereby patients with null mutations bear a risk of >75%, compared to a risk of <10% in patients with missense mutations or small F8 deletions and insertions [Citation27–Citation30].
The strong relationship between F8 genotype and the risk of inhibitor development is best explained by the lack of central immune tolerance to FVIII in the hemophilia A patients with null mutations, such as large deletions [Citation11,Citation31]. Due to the complete absence of autologous FVIII in these patients, FVIII-reactive T- and B-cells are not eliminated in their ontogeny in the thymus or bone marrow and could become activated after FVIII administration. However, the fact that not all patients with severe hemophilia A develop inhibitors, implies that also peripheral immune tolerance mechanisms play a role in preventing pathologic manifestations of FVIII-reactive T- and B-cells. These mechanisms include, for example, the presentation of self-antigens as peptide/MHC complexes by DCs in steady state to result in anergy or deletion of autoreactive T- or B-cell clones and the induced suppressive activity of regulatory T-cells (Tregs) [Citation12,Citation31,Citation32].
Moreover, there are several requirements for an effective interaction between the APC and the T-cell in order to generate an alloantibody response to FVIII treatment [Citation12,Citation15]. This consists of the functional avidity of the particular T-cell to bind to the MHC-peptide complex and, as stated before, the up-regulation of co-stimulatory signals on the APC in the presence of danger signals and the release of pro-inflammatory cytokines. Absence of these costimulatory signals results in a non-productive immunologic synapse between the APC and the T-cell, which prevents T-cells to become fully activated and likely inhibits T-cell effector function () [Citation31–Citation33].
Contrary to the well-established relationship between FVIII mutation and inhibitor risk, this association is less clear for environmental factors, including the FVIII product type as potential risk factor. Until recently, studies regarding this issue showed contradictory and/or inconclusive results [Citation34–Citation36]. The SIPPET study was specifically designed to answer this question. It is the first and only randomized controlled trial showing an increased inhibitor risk by recombinant FVIII (rFVIII) products compared to plasma-derived FVIII (pd-FVIII) [Citation37]. This is supported by a French national cohort study [Citation24]. Proposed explanations for this finding include the presence of Von Willebrand Factor (VWF) in pd-FVIII, reducing FVIII immunogenicity by epitope masking and prevention of endocytosis by DCs. Secondly, the different posttranslational modifications of rFVIII due to its production in mammalian cells rather than human cells might influence its immunogenicity [Citation38–Citation42]. This issue however remains highly debated, which is beyond the scope of this review.
Taken together, there are many factors involved in the delicate equilibrium between tolerance and immunization to FVIII. As proposed by a model of van Helden et al. genetic factors set the individual threshold for inhibitor development [Citation43]. Subsequent environmental factors, such as intensity of treatment at first exposure, FVIII product type and administration of FVIII in the absence or presence of inflammation, determine whether the immune activation exceeds the immune threshold for inhibitor formation. Although a simplified representation of the complexity of the immune response to FVIII, this model provides better understanding of why only a fraction of all patients with hemophilia A develops inhibitors. Intriguing in this model is the influence of danger signals in the break of tolerance. The next part of this review will focus on this issue, starting with a general explanation of the ‘danger theory’, followed by evidence from both pre-clinical and clinical studies of this theory in inhibitor development in hemophilia A.
3. Danger theory
The danger theory, as proposed by Polly Matzinger in 1994, suggests that the immune system’s primary task is to detect and protect our body against danger [Citation10,Citation44]. This theory strongly opposes the classical concept of the immune system that the main concern of our immune cells is to discriminate between non-self and self, protecting our body against the first while maintaining tolerance towards the latter.
This so-called self-/non-self discrimination (SNSD) model started by Burnet in 1959. He proposed that each B-cell carries a specific receptor that recognizes foreign antigens and that self-reactive lymphocytes are deleted early in life [Citation45,Citation46]. Since then this model is modified several times. First of all, it was recognized that B-cells require help from T-helper cells in order to get activated [Citation47]. Moreover, it became evident that only the recognition of (foreign) antigens, i.e. signal 1, was not sufficient to elicit a proper T-cell response, and that a second signal, signal 2, in the form of co-stimulation from antigen presenting cells (APCs) was necessary. Absence of this signal 2 would lead to tolerance of the T-cell. This finding emphasizes the crucial role of the innate immune system in contributing to the activation of the adaptive immune response.
In 1989, Charles Janeway Jr. introduced the infectious non-self theory of immunity, stating that the innate system acts as a sensor of pathogenic invasion by the means of evolutionarily conserved receptors called pattern recognition receptors (PRRs), for example the Toll-like receptors (TLRs) and NOD-like receptors [Citation48,Citation49]. These receptors are able to recognize and bind to conserve molecular constituents, also known as PAMPs [Citation48,Citation49]. Although the discovery of these PRRs and PAMPs explained how the (innate) immune system could differentiate between infectious and non-infectious antigens, still many questions remained unanswered [Citation50]. For example, why are the non-self commensal bacteria in our gut or is the fetus not attacked by its mother? Why is it necessary to add an adjuvant to our vaccines and how do nonbacterial adjuvants work? Moreover, during life ‘self’ changes, how does our immune system deal with this changing ‘self’?
In order to answer these questions, Matzinger conceived the danger model, which challenged the central paradigm in immunology by claiming that the immune system is more concerned with damage than with foreignness and that immune cells become activated by alarm signals from injured tissues rather than the recognition of non-self [Citation10,Citation44]. Hereby the context in which the antigen contact takes place, dictates the immune response. Local alarm or danger signals from this micro-environment activate APCs, which in turn are able to induce T-cells by providing the necessary co-stimulation. These danger signals can arise from both exogenous (PAMPs) as well as endogenous sources. The endogenous ‘alarmins’ are mostly associated with host cells in distress and necrotic cell death [Citation51]. These so-called DAMPs include heat-shock proteins, uric acid, and DNA associated proteins like high mobility group box 1 (HMGB1) [Citation52–Citation55]. As proposed by Seong and Matzinger, PAMPs and DAMPs share similar conserved hydrophobic portions on their respective molecules, which enable them to bind to the same PRRs and therefore to induce comparable inflammatory responses [Citation56]. Importantly these endogenous and exogenous alarm molecules are not necessary directly related to the antigen, which could explain why the immune system not only reacts to ‘dangerous’ non-self infectious agents, like bacteria, but also to self-antigens from healthy cells during autoimmune diseases or to FVIII as is the case with inhibitor development in hemophilia A.
With regard to inhibitor development in hemophilia A, several danger signals have been proposed. These include exogenous triggers such as infections and vaccinations that occur around the time of treatment with FVIII on one side, whereas trauma, surgery and severe bleeds are considered as the most important endogenous danger signals [Citation57,Citation58]. All these events are associated with necrotic cell damage, resulting in a local micro-environment now harboring danger signals. Subsequently, this environment could now support the generation of immune reactivity, setting the stage to produce neutralizing antibodies to the administered FVIII (). Importantly, with the same reasoning it is proposed that administration of FVIII in the absence of danger signals, as is the case with prophylactic treatment, would promote tolerance to the deficient coagulation protein.
To conclude, theoretically spoken the presence of danger signals during FVIII administration could contribute to the risk of inhibitor formation. The next part of this review will focus on the evidence for this hypothesis, based on both pre-clinical and clinical studies.
4. Preclinical data
Testing the danger theory in a preclinical setting is attractive, because in-patient studies for ethical and practical reasons are difficult to clarify the impact of a single factor in the complex etiology of inhibitor development (). Nonetheless, in vitro the challenge is to simulate the dynamic interplay of the different immunological processes involved. Animal models can provide this complex immunologic environment, but here it is important to realize that human FVIII infused in knockout mice or rats, is a xenoprotein with a homology of only 51% compared to rat FVIII [Citation59]. Novel transgenic mouse models with human MHC II expression are generated to study inhibitor development and tolerance [Citation60,Citation61].
Table 1. Summary of preclinical and clinical observations related to the danger theory.
4.1. In vitro and basic immunological in vivo studies
One approach of in vitro studies to test the danger theory involves the stimulation of DCs from healthy donors with FVIII in the presence of the bacterial danger signal lipopolysaccharide (LPS). Such treatment results in an increased expression of co-stimulatory molecules CD80, CD83 and CD86 on DCs as compared to FVIII or LPS alone [Citation62]. The intensity of this response is dependent on the CD86 expression on DCs before stimulation as well as on the type and amount of co-applied danger signal. LPS is one example of a danger signal, but other bacterial danger signals, and synthetic nucleic acids exist that are able to increase CD83 and CD86 expression on DCs. Such innate immune activation is necessary to establish adaptive immune responses, including inhibitor production. The synergistic DC activation results in an increased proliferation of CD4+ memory T-cells [Citation63]. Co-stimulation via CD86 and adequate antigen presentation on MHC class II molecules are a prerequisite for this CD4+ activation. Taken together, these data demonstrate that FVIII plus danger signals synergistically increase DC activation and subsequent CD4 + T-cell responses, supporting the danger theory. An important limitation is the use of healthy controls instead of hemophilia A patients.
Given the critical role of FVIII within the coagulation cascade and the association between coagulation and inflammation, the question whether the FVIII protein itself could act as a danger signal has been evaluated as well [Citation64,Citation65]. In a study of Pfistershammer et al. it was shown that neither FVIII, thrombin-activated FVIII, nor FVIII-VWF complex modulates the maturation of human dendritic cells (DCs) or their ability to stimulate T cells [Citation66]. Therefore, the authors concluded that neither of these proteins act as a danger signal to human DCs [Citation66]. The possibility that FVIII itself acts not directly as danger signal, but indirectly by the formation of other coagulation proteins, especially thrombin, has been evaluated as well. Skupsky et al. demonstrated in a hemophilia A mouse model that both coadministration of FVIII and ovalbumin (OVA) as well as thrombin and OVA induced immune responses to OVA, which by itself is poorly immunogenic [Citation67]. Moreover, warfarin and the direct thrombin inhibitor, hirudin, significantly reduced B- and T-cell response to FVIII [Citation67]. However, two other hemophilia A mice studies did not support the hypothesis that thrombin generation or coagulation-associated processes modulate the anti-FVIII antibody response [Citation68,Citation69]. In one of these studies, both inhibition of tissue factor activation and inhibition of the coagulation did not modulate the immune response to FVIII [Citation69]. Moreover, no significant differences in the immunogenicity were found between wildtype B domain-deleted FVIII and an inactive FVIII molecule (V634M FVIII) [Citation68,Citation69].
Taken together, so far evidence that FVIII, by itself or its procoagulant function, could act as a danger signal is lacking.
4.2. In vivo studies investigating clinical danger signals
Different danger signals are tested in rodent models: surgery, haemarthrosis, and vaccination. These studies are described in the next paragraphs and summarized in .
4.2.1. Surgery
In trauma and surgery cell damage leads to the release of endogenous danger signals, or DAMPs, activating the immune system, in addition to the requirement of intensive treatment with high doses of FVIII supplementation. As such, it is difficult to assess the contribution of surgery or trauma to the development of inhibitors apart from the effect of peak treatment. In a murine study, a potential contribution of endogenous danger signals to increased inhibitor formation could not be confirmed [Citation70]. In this study, laparotomy was performed and caused a detectable inflammatory response, yet this was not associated with an increase in FVIII inhibitor development. Factor VIII knock-out mice were used with or without humanization of MHC II molecules. All mice without a humanized MHC II developed high-titer antibodies and there was no difference in the antibody titer, whereas in the mice with a humanized MHC II only a proportion developed detectable antibodies without a difference between surgery and control groups in terms of proportion and titer. However, when FVIII administration was combined with LPS in immunologically tolerant mice, inhibitors developed in 100% of cases.
4.2.2. Haemarthrosis
Joint bleeds also cause tissue damage and inflammation, require FVIII replacement therapy and thereby increase risk to inhibitor formation. The effect of a joint bleed on inhibitor development is investigated in two animal studies with contradictory results. In a murine study, the danger theory was not confirmed [Citation71]. A joint bleed was induced by needle puncture, and factor VIII replacement therapy was once weekly was started either 1 or 14 days after injury. There was no increase in the anti-FVIII antibody production in mice compared to uninjured mice. Importantly, the rate of inhibitor development was 100% in all groups and extremely high inhibitor levels were detected, also in uninjured mice. This experimental setup prevented addressing a possible stimulatory effect of joint bleeding in the FVIII-specific antibody response.
In contrast, a needle-induced joint bleed did significantly increase the inhibitor response to human FVIII in hemophilia A rats compared to sham-operated rats (80% versus 33%, respectively) [Citation59]. The inhibitor levels were significantly higher after a knee bleed. These data show that in rats FVIII replacement at the time of a joint bleed potentiates inhibitor development, and this is possibly due to the systemic inflammatory response induced by the bleed [Citation72]. In human, similar such controlled studies are yet to be reported.
4.2.3. Vaccination and infection
Antigen administration in combination with an adjuvant to stimulate the immune system is fundamental to successful vaccination. As such, a vaccination might act as a danger signal. The same applies for infections. The effect of influenza vaccinations given intramuscularly (i.m.) or intravenously (i.v.) prior to multiple infusions of FVIII was tested in a mouse model of hemophilia A [Citation73]. Surprisingly, this study suggested a protective effect of i.m. vaccination to inhibitor development, but not by i.v. vaccination. It was hypothesized that the reduced anti-FVIII antibody response after i.m. vaccination was due to antigen competition, by means of lymphocyte recruitment to the immunization site. However, no definitive conclusions can be drawn from this preclinical study yet.
5. Clinical observations
Clinical studies investigating the impact of a single factor in inhibitor formation are challenging as many factors affect outcome and hemophilia is a rare disease. Data mainly come from large retrospective cohort studies or small case–control studies ().
5.1. Surgery
In 2000 two patients with mild hemophilia developed high-titer inhibitors after continuous infusion with recombinant FVIII as prophylaxis for surgery, suggesting a role for intensive treatment in inhibitor development at the time of surgery [Citation74]. This was confirmed by a case series of 54 boys with mild hemophilia showing inhibitor formation in four patients after continuous infusion (for surgery, haemarthrosis and intracranial bleeding) [Citation75] and in a cohort study of 138 mild/moderate hemophilia A patients where 7 patients developed an inhibitor after intensive peri-operative replacement therapy [Citation76]. Interestingly, in the latter study intensive FVIII replacement therapy for bleeding was not associated with inhibitor development, whereas the inhibitor risk in peri-operatively treated patients was severely increased, especially in patients with the Arg593Cys mutation.
In previously untreated patients (PUPs) with severe hemophilia, peak treatment at the time of surgery is related to an increased inhibitor risk as well. Gouw et al. combined data from 236 patients included in four recombinant FVIII PUP studies, showing a 1.6 increased relative risk for peak treatment moments and 2.7 for surgical procedures [Citation77]. This was confirmed by the CANAL study, a large observational PUP cohort study. The inhibitor risk was 3.7 and 3.3 times increased for peak treatment and surgery, respectively, [Citation22]. In this study, the risk of inhibitor development was markedly higher when patients were treated for surgical procedures (65%) than patients who were first treated for bleeds or prophylactically (23% and 22%, respectively). Major surgeries at the first treatment episode most significantly increased the risk of developing inhibitors, although surgeries at any exposure day were associated with an increased risk. Also in the RODIN study, intensive FVIII treatment of surgery other than implantation of venous access devices was associated with an 1.4 to 2.0 times increased inhibitor risk, although this was not statistically significant [Citation4]. Also in a more recent study in 825 PUPs, surgery as the reason for first exposure was associated with a 2.4 odds ratio for inhibitor development, whereas a bleed was not [Citation78].
However, not all studies confirm this association. A case–control study by Santagostino et al. did not find a higher prevalence of surgery among 60 inhibitors compared with 48 non-inhibitor patients [Citation79]. A major limitation is the size of the study in which selection bias could not be ruled out. The number of surgeries was low, 15 (25%) surgeries in the cases and 11 (23%) in the controls. And although not statistically significant, the number of cases with surgery at first FVIII infusion was 7 (12%) in the cases compared to only 2 (4%) in the controls. Moreover, a retrospective case–control study by Maclean et al. did not find an increased risk of inhibitor development by surgery in 39 patients, neither in 15 patients (nine controls and six cases) receiving intensive treatment for 5 days [Citation80].
For previously treated patients (PTPs, after at least 50 exposure days), different prospective and retrospective cohort studies demonstrate that intensive therapy at the time of surgery, also when given as continuous infusion, does not increase inhibitor risk [Citation57].
5.2. (Joint) bleeds
Studies regarding haemarthrosis as a risk factor for inhibitor development are conflicting. Peak treatment at the time of a bleed was associated with an increased inhibitor risk in both PUP cohorts mentioned in the previous paragraph [Citation4,Citation22]. This was most prominent when treatment was given on at least 3–5 consecutive days immediately at the first treatment episode. This was also described by Vézina et al. in a PUP cohort where haemarthrosis as the indication for first exposure to FVIII treatment was associated with an 7.63 increased risk of inhibitor development [Citation81]. Maclean et al. found a 4.1 time increased the risk of inhibitor development in patients treated for life-threatening bleeds (11 patients and 3 controls) [Citation80].
In contrast, in the study by Santagostino et al. the risk was not increased by treatment at the time of bleeds in PUPs and minimally treated patients [Citation79], neither was a bleed as a reason for first exposure in a large PUP cohort [Citation78]. Also in a cohort of mild hemophilia patients, peak treatment for bleeds was not associated with inhibitor development. In total 22 patients received their first intensive FVIII concentrate administration for a bleeding episode and none developed an inhibitor. Compared to 41 patients receiving intensive treatment for surgery which was associated with an increased inhibitor risk (see above), there was no difference in the number of exposure days (EDs) preceding the intensive treatment period nor in the number of EDs during intensive treatment nor the total dose of FVIII concentrate administered [Citation76]. Thus, studies on the impact of joint bleeds on inhibitor development failed to demonstrate a clear association.
5.3. Vaccination and infection
No apparent association with the development of inhibitors and the frequency of FVIII during infections or vaccinations was found in the previously mentioned case–control study by Santagostino [Citation79]. It is important to realize that the number of patients with an infection or vaccinations during active FVIII treatment was low with only 12 patients and 11 controls. In the case–control study by Maclean, concomitant infection was not found to be significantly associated with inhibitor development (OR: 1.8, CI: 0.5–6.5) [Citation80]. Also, McMillan et al. did not find a relationship between inhibitor development and illnesses occurring within the preceding year [Citation82].
Recently, retrospective data from the PedNet Registry investigating 375 PUPs with severe hemophilia who had received vaccinations within the first 75 exposure days, also rejected the hypothesis that vaccinations given in close proximity to FVIII exposure are associated with inhibitor development [Citation83].
5.4. Prophylaxis
The aim of early prophylactic treatment is to prevent major bleeds. By administrating FVIII in the absence of danger signals, immune tolerance might be induced. As such, early prophylaxis could play a protective role in inhibitor development. Several studies evaluated this hypothesis.
In the retrospective CANAL cohort study, it was shown that regular prophylaxis was associated with a lower inhibitor risk compared to on-demand therapy [Citation77]. A case–control study of the UKHCDO could not confirm this finding [Citation80]. In the prospective RODIN study, prophylactic treatment was associated with a decreased inhibitor risk after 20 exposure days (EDs) of FVIII and this correlation was more pronounced in patients with low-risk F8 genotypes compared to high-risk F8 genotypes [Citation4].
Kurnik et al. performed a pilot study to evaluate whether low-dose prophylaxis for the first 20–50 EDs in combination with avoidance of immunological dangers signals could induce tolerance and minimize the incidence of inhibitors [Citation84]. In this study, an early prophylaxis regimen was associated with a significant lower risk of inhibitor development compared to patients treated with a standard prophylaxis regimen. In order to verify the protective effect of early prophylaxis and minimization of immunological danger signals, the prospective EPIC (Early Prophylaxis Immunologic Challenge) study was designed [Citation85]. Contrary to the authors’ hypothesis, this study had to be terminated prematurely due to an excess inhibitor incidence (8/19 patients, 42%).
Thus, the protective effect of prophylaxis on inhibitor development by avoiding danger signals during FVIII administration is still not fully elucidated.
6. Conclusion
Inhibitor development is a multifactorial process caused by the complex interplay between genetic and environmental factors. Dissecting the contribution of a single factor like surgery or joint bleeding is difficult as these events are accompanied by peak treatment, which also is a risk factor for inhibitor development by itself. In vitro studies are scarce and to the best of our knowledge only performed in healthy controls. Although the results from these studies support the danger theory, translating these in vitro data to clinical practice in hemophilia A is challenging. For the preclinical studies, the number of relevant studies is limited and it is important to realize that the inhibitor rate in general is high. The infused clotting factor is perceived by the immune system as a foreign protein and as such it is hard to demonstrate a contributing effect of a danger signal. In a recent published paper, it even proposes that all hemophilia A patients develop anti-FVIII immune responses regardless the presence of danger signals and that particularly the ability to develop counteractive tolerogenic reactions determines if a patients will develop clinically significant inhibitors or not [Citation86].
Clinical data from cohort studies and case–control studies show that surgery at first FVIII exposure is a risk factor for inhibitor development, whereas for surgery at subsequent exposures its risk seems less clear. Evidence for (joint) bleeds as a potential danger signal is still inconclusive. For vaccinations and infections, although based on scarce evidence, the contribution to inhibitor development seems absent.
In summary, although the danger model provides a theoretical background why and how danger signals could contribute to inhibitor development in hemophilia A, the data to support this hypothesis are scarce and not conclusive.
7. Expert opinion
Inhibitor development is the biggest challenge in hemophilia A treatment at present and we are still unable to predict the patients’ individual risk. As hemophilia A is a rare disease and inhibitor development is an intricate multifactorial process, it is difficult to unravel the contribution of a single factor like danger signals in clinical studies, or to incorporate all factors in in vitro or in vivo model-based studies. Data from large, preferably prospective, multinational registry studies are most useful as they offer the possibility to collect data from a relatively large number of patients. This is especially important as the treatment landscape in hemophilia is rapidly changing. It will remain challenging to distinguish the contribution of peak treatment from the danger signal, but it is the question whether this is necessary as these risk factors are almost always combined. Eventually, such registry studies might lead to individual prediction models and further guidance for treatment, for example to avoid surgeries, start early prophylaxis or to choose non-replacement strategies.
The treatment landscape in hemophilia A is changing rapidly, with the introduction of non-factor replacement strategies like a bispecific antibody mimicking the cofactor activity of FVIII or approaches inhibiting natural anticoagulants such as tissue factor pathway inhibitor (TFPI) and antithrombin [Citation87–Citation89]. This provides new therapeutic options for patients with inhibitors, but also may prevent inhibitor development. However, in theory the development of antibodies against these agents is possible. Also, replacement therapy will still be used as it is very effective and the possibility to monitoring FVIII levels is an important advantage compared to non-factor replacement strategies.
Tools to predict the change of inhibitor development based on data from large registries might guide treatment choices in the near future.
Other interesting experimental approaches to prevent inhibitor development, especially with regard to the danger theory, include administration of FVIII combined with antibodies that block costimulatory pathways. Potential targets herein are CD28 and CD80/CD86, inducible T-cell costimulatory (ICOS) and CD40 and CD40L. Animal models show that administration of these antibodies is associated with induction of Tregs and the prevention of inhibitor formation in naïve hemophilia A mice, although the duration of this effect appears limited [Citation90–Citation93]. Translation of these strategies from preclinical studies to the clinical practice of hemophilia A might improve future patient care.
Article highlights
Inhibitor development is a complex and multifactorial process, involving both genetic and environmental factors.
Danger signals might contribute to inhibitor development; however, the evidence is scarce and not conclusive.
Future studies elucidating the role of danger signals, like multinational registries, are warranted, but challenging.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Carcao M. The diagnosis and management of congenital hemophilia. Semin Thromb Hemost [Internet]. 2012;38:727–734. Available from: http://www.thieme-connect.de/DOI/DOI?10.1055/s-0032-1326786
- Tabriznia-Tabrizi S, Gholampour M, Mansouritorghabeh H. A close insight to factor VIII inhibitor in the congenital hemophilia A. Expert Rev Hematol [Internet]. 2016;9:903–913.
- Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9:418–435.
- Gouw S, van Den Berg H, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood [Internet]. 2013;121:4046–4055. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23553768
- Walsh CE, Jimenez-Yuste V, Auerswald G, et al. The burden of inhibitors in haemophilia patients. Thromb Haemost. 2016;116:S10–7.
- Gringeri A, Mantovani LG, Scalone L, et al. Cost of care and quality of life for patients with hemophilia complicated by inhibitors : the COCIS study group. Blood. 2003;102:2358–2363.
- Hay CRM, Dimichele DM. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119:1335–1344.
- Valentino LA, Kempton CL, Kruse-Jarres R, et al. US guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia. 2015;21:559–567.
- Rocino A, Cortesi PA, Scalone L, et al. Immune tolerance induction in patients with haemophilia a and inhibitors: effectiveness and cost analysis in an European Cohort (The ITER Study). Haemophilia. 2016;22:96–102.
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045.
- White GC, Kempton CL, Grimsley A, et al. Cellular immune responses in hemophilia: why do inhibitors develop in some, but not all hemophiliacs? J Thromb Haemost. 2005;3:1676–1681.
- Lacroix-Desmazes S, Navarrete AM, André S, et al. Dynamics of factor VIII interactions determine its immunologic fate in hemophilia A. Blood. 2008;112:240–249.
- Waters B, Lillicrap D. The molecular mechanisms of immunomodulation and tolerance induction to factor VIII. J Thromb Haemost. 2009;7:1446–1456.
- Wroblewska A, Reipert BM, Pratt KP, et al. Dangerous liaisons : how the immune system deals with factor VIII. J Thromb Haemost. 2013;11:47–55.
- Georgescu MT, Lai JD, Hough C, et al. War and peace: factor VIII and the adaptive immune response. Cell Immunol. 2016;301:2–7.
- Herczenik E, van Haren SD, Wroblewska A, et al. Uptake of blood coagulation factor VIII by dendritic cells is mediated via its C1 domain. J Allergy Clin Immunol [Internet]. 2012;129:501–509.e5.
- Wroblewska A, van Haren SD, Herczenik E, et al. Modification of an exposed loop in the C1 domain reduces immune responses to factor VIII in hemophilia A mice. Blood. 2012;119:5294–5300.
- Dasgupta S, Navarrete A-M, Bayry J, et al. A role for exposed mannosylations in presentation of human therapeutic self-proteins to CD4+ T lymphocytes. Proc Natl Acad Sci U S A. 2007;104:1–6.
- André S, Meslier Y, Dimitrov JD, et al. A cellular viewpoint of anti-FVIII immune response in hemophilia a. Clin Rev Allergy Immunol. 2009;37:105–113.
- Navarrete A, Dasgupta S, Delignat S, et al. Splenic marginal zone antigen-presenting cells are critical for the primary allo-immune response to therapeutic factor VIII in hemophilia A. J Thromb Haemost. 2009;7:1816–1823.
- Peyvandi F, Ettingshausen CE, Goudemand J, et al. New findings on inhibitor development: from registries to clinical studies. Haemophilia [Internet]. 2017;23:4–13.
- Gouw SC, van der Bom JG, van Den Berg HM. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007;109:4648–4654.
- Peyvandi F, Garagiola I. Product type and other environmental risk factors for inhibitor development in severe hemophilia A. Res Pract Thromb Haemost. 2018;2:220–227.
- Calvez T, Chambost H, D’Oiron R, et al. Analyses of the FranceCoag cohort support differences in immunogenicity among one VIII brands in boys with severe hemophilia A. Haematologica. 2018;103:179–189.
- Garagiola I, Palla R, Peyvandi F. Risk factors for inhibitor development in severe hemophilia A. Thromb Res [Internet]. 2018;168:20–27.
- Repessé Y, Peyron I, Dimitrov JD, et al. Development of inhibitory antibodies to therapeutic factor VIII in severe hemophilia A is associated with microsatellite polymorphisms in the HMOX1 promoter. Haematologica. 2013;98:1650–1655.
- Schwaab R, Brackmann H, Meyer C, et al. Haemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost. 1995;74:1402–1406.
- Gouw S, van der Bom J, van Den Berg H, et al. Influence of the type of F8 gene mutation on inhibitor development in a single centre cohort of severe haemophilia A patients. Haemophilia. 2011;17:275–281.
- Goodeve A, Peake I. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Hemost. 2003;29:23–30.
- Lillicrap D. The role of immunomodulation in the management of factor VIII inhibitors. Hematol Am Soc Hematol Educ Progr [Internet]. 2006:421–425. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17124093
- Anderton SM, Wraith DC. Selection and fine-tuning of the autoimmune T-cell repertoire. Nat Rev Immunol [Internet]. 2002;2:487–498. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12094223
- Walker LSK, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol [Internet]. 2002;2:11–19. Available from: http://www.nature.com/doifinder/10.1038/nri701
- Bluestone J, Abbas A. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–257.
- Iorio A, Halimeh S, Holzhauer S, et al. Rate of inhibitor development in previously untreated hemophilia A patients treated with plasma-derived or recombinant factor VIII concentrates: A systematic review. J Thromb Haemost. 2010;8:1256–1265.
- Franchini M, Coppola A, Rocino A, et al. Systematic review of the role of FVIII concentrates in inhibitor development in previously untreated patients with severe hemophilia A: a 2013 update. Semin Thromb Hemost. 2013;39:752–766.
- Marcucci M, Mancuso MEL, Santagostino E, et al. Type and intensity of FVIII exposure on inhibitor development in PUPs with haemophilia A. A patient-level meta-analysis. Thromb Haemost. 2015;113:958–967.
- Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med [Internet]. 2016;374:2054–2064. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27223147%5Cnhttp://www.nejm.org/doi/10.1056/NEJMoa1516437
- Mannucci PM, Shi Q, Bonanad S, et al. Novel investigations on the protective role of the FVIII/VWF complex in inhibitor development. Haemophilia. 2014;20:2–16.
- Franchini M, Lippi G. Von Willebrand factor-containing factor VIII concentrates and inhibitors in haemophilia A: A critical literature review. Thromb Haemost. 2010;104:931–940.
- Di Minno G, Coppola A. A role for von Willebrand factor in immune tolerance induction in patients with haemophilia A and inhibitors? Blood Transfus [Internet]. 2011;9 Suppl 2:s14–s20. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3159912&tool=pmcentrez&rendertype=abstract
- Hironakaso T, Furukawas K, Fournelll MA, et al. Comparative study of the sugar chains of factor VIII purified from human plasma and from the culture media of recombinant baby hamster kidney cells. J Biol Chem. 1992;267:8012–8020.
- Kannicht C, Ramström M, Kohla G, et al. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thromb Res [Internet]. 2013;131:78–88.
- van Helden PMW, van Haren SD, Fijnvandraat K, et al. Factor VIII-specific B cell responses in haemophilia A patients with inhibitors. Haemophilia. 2010;16:35–43.
- Matzinger P. The danger model : a renewed sense of self. Science. 2002;296:301–306.
- Fekety FR. The Clonal Selection Theory of Acquired Immunity. Yale J Biol Med. 1960 Jun;32(6):480.
- Burnet F. A modification of Jerne’s theory of antibody production using the concept of clonal selection. CA Cancer J Clin. 1976;26:119–121.
- Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. 1970;69:1042–1049.
- Janeway C. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992;13:11–16.
- Medzhitov R, Janeway C. How does the immune system distinguish self from nonself? Semin Immunol. 2000;12:185–188.
- Matzinger P. Essay 1 : the danger model in its historical context. Scand J Immunol. 2001;54:4–9.
- Kono H, Rock K. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289.
- Shi Y, Evans J, Rock K. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521.
- Lotze M, Tracey K. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342.
- Bianchi M. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5.
- Hwang PF, Porterfield N, Pannell D, et al. Trauma is danger. J Transl Med. 2011;9:1–10.
- Seong S, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478.
- Astermark J, Altisent C, Batorova A, et al. Non-genetic risk factors and the development of inhibitors in haemophilia: a comprehensive review and consensus report. Haemophilia. 2010;16:747–766.
- Lövgren KM, Sondergaard H, Skov S, et al. Non-genetic risk factors in haemophilia A inhibitor management – the danger theory and the use of animal models. Haemophilia. 2016;22:657–666.
- Lövgren K, Søndergaard H, Skov S, et al. Antibody response to recombinant human coagulation factor VIII in a new rat model of severe hemophilia A. J Thromb Haemost. 2016;14:747–756.
- Reipert B, Arruda V, Lillicrap D. Animal models of inhibitors. Haemophilia. 2010;16:47–53.
- Lillicrap D, Fijnvandraat K, Santagostino E. Inhibitors - genetic and environmental factors. Haemophilia. 2014;20:87–93.
- Miller L, Weissmüller S, Ringler E, et al. Danger signal-dependent activation of human dendritic cells by plasma-derived factor VIII products. Thromb Haemost. 2015;114:268–276.
- Miller L, Ringler E, Kistner K, et al. Human dendritic cells synergistically activated by FVIII plus LPS induce activation of autologous CD4+ T cells. Thromb Haemost. 2018;118:688–699.
- Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417–430.
- Delvaeye M, Conway EM. Coagulation and innate immune responses: can we view them separately? Blood. 2009;114:2367–2374.
- Pfistershammer K, Stöckl J, Siekmann J, et al. Recombinant factor VIII and factor VIII-von Willebrand factor complex do not present danger signals for human dendritic cells. Thromb Haemost. 2006;96:309–316.
- Skupsky J, Zhang AH, Su Y, et al. A role for thrombin in the initiation of the immune response to therapeutic factor VIII. Blood. 2009;114:4741–4748.
- Meeks SL, Cox CL, Healey JF, et al. A major determinant of the immunogenicity of factor VIII in a murine model is independent of its procoagulant function. Blood. 2012;120:2512–2520.
- Gangadharan B, Delignat S, Ollivier V, et al. Role of coagulation-associated processes on factor VIII immunogenicity in a mouse model of severe hemophilia A. J Thromb Haemost. 2014;12:2065–2069.
- Moorehead P, Waters B, Sponagle K, et al. Surgical injury alone does not provoke the development of factor VIII inhibitors in mouse models of hemophilia A. Blood. 2012;120:627.
- Peyron I, Dimitrov J, Delignat S, et al. Haemarthrosis and arthropathy do not favour the development of factor VIII inhibitors in severe haemophilia A mice. Haemophilia. 2015;21:e94–8.
- Lövgren K, Christensen K, Majewski W, et al. Acute haemarthrosis in the haemophilia A rat generates a local and systemic proinflammatory response. Thromb Haemost. 2017;117:2092–2104.
- Lai J, Moorehead P, Sponagle K, et al. Concurrent influenza vaccination reduces anti-FVIII antibody responses in murine hemophilia A. Blood. 2016;127:3439–3449.
- White B, Cotter M, Byrne M, et al. High responding factor VIII inhibitors in mild haemophilia - Is there a link with recent changes in clinical practice? Haemophilia. 2000;6:113–115.
- Sharathkumar A, Lillicrap D, Blanchette V, et al. Intensive exposure to factor VIII is a risk factor for inhibitor development in mild hemophilia A. J Thromb Haemost. 2003;1:1228–1236.
- Eckhardt C, Menke L, van Ommen C, et al. Intensive peri-operative use of factor VIII and the Arg593 → Cys mutation are risk factors for inhibitor development in mild/moderate hemophilia A. J Thromb Haemost. 2009;7:930–937.
- Gouw S, van Den Berg H, Le Cessie S, et al. Treatment characteristics and the risk of inhibitor development: A multicenter cohort study among previously untreated patients with severe hemophilia A. J Thromb Haemost. 2007;5:1383–1390.
- Hashemi S, Fischer K, Moons K, et al. Improved prediction of inhibitor development in previously untreated patients with severe haemophilia A. Haemophilia. 2015;21:227–233.
- Santagostino E, Mancuso M, Rocino A, et al. Environmental risk factors for inhibitor development in children with haemophilia A: a case-control study. Br J Haematol. 2005;130:422–427.
- Maclean P, Richards M, Williams M, et al. Treatment related factors and inhibitor development in children with severe haemophilia A. Haemophilia. 2011;17:282–287.
- Vézina C, Carcao M, Infante-Rivard C, et al. Incidence and risk factors for inhibitor development in previously untreated severe haemophilia A patients born between 2005 and 2010. Haemophilia. 2014;20:771–776.
- McMillan C, Shapiro S, Whitehurst D, et al. The natural history of factor VIII:C inhibitors in patients with hemophilia A: a national cooperative study. II. Observations on the initial development of factor VIII:C inhibitors. Blood. 1988;71:344–348.
- Platokouki H, Fischer K, Gouw S, et al. Vaccinations are not associated with inhibitor development in boys with severe haemophilia A. Haemophilia. 2018;24:283–290.
- Kurnik K, Bidlingmaier C, Engl W, et al. New early prophylaxis regimen that avoids immunological danger signals can reduce FVIII inhibitor development. Haemophilia. 2010;16:256–262.
- Auerswald G, Kurnik K, Aledort LM, et al. The EPIC study: A lesson to learn. Haemophilia. 2015;21:622–628.
- Varthaman A, Lacroix-Desmazes S. Pathogenic immune response to therapeutic factor VIII: exacerbated response or failed induction of tolerance? Haematologica. 2019;104:236–244.
- Oldenburg J, Mahlangu J, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med [Internet]. 2017;377:809–818.
- Chowdary P, Lethagen S, Friedrich U, et al. Safety and pharmacokinetics of anti-TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: A randomized first human dose trial. J Thromb Haemost. 2015;13:743–754.
- Pasi K, Rangarajan A, Georgiev P, et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med [Internet]. 2017;377:819–828.
- Qian J, Collins M, Sharpe AH, et al. Prevention and treatment of factor VIII inhibitors in murine hemophilia A. Blood [Internet]. 2000;95:1324–1329. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10666206%5Cnhttp://bloodjournal.org/content/bloodjournal/95/4/1324.full.pdf%5Cnhttp://www.bloodjournal.org/content/bloodjournal/95/4/1324.full.pdf
- Reipert BM, Sasgary M, Ahmad RU, et al. Blockade of CD40/CD40 ligand interactions prevents induction of factor VIII inhibitors in hemophilic mice but does not induce lasting immune tolerance. Thromb Haemost. 2001;86:1345–1352.
- Rossi G, Sarkar J, Scandella D. Long-term induction of immune tolerance after blockade of CD40-CD40L interaction in a mouse model of hemophilia A. Blood. 2001;97:2750–2757.
- Hausl C, Ahmad RU, Schwarz HP, et al. Preventing restimulation of memory B cells in hemophilia A: A potential new strategy for the treatment of antibody-dependent immune disorders. Blood. 2004;104:115–122.
- Astermark J, Berntorp E, White GC, et al. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development. Haemophilia. 2001;7:267–272.
- Hay C, Ollier W, Pepper L, et al. HLA class II profile: a weak determinant of factor VIII inhibitor development in severe haemophilia A. UKHCDO inhibitor working party. Thromb Haemost. 1997;77:234–237.
- Astermark J, Oldenburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1beta and IL4 genes are associated with inhibitor development in patients with hemophilia A. Blood. 2006;107:3167–3172.
- Astermark J, Wang X, Oldenburg J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in patients with hemophilia A. J Thromb Haemost [Internet]. 2007;5:263–265. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17269936
- Pavlova A, Delev D, Lacroix-Desmazes S, et al. Impact of polymorphisms of the major histocompatibility complex class II, interleukin-10, tumor necrosis factor-α and cytotoxic T-lymphocyte antigen-4 genes on inhibitor development in severe hemophilia A. J Thromb Haemost. 2009;7:2006–2015.
- Chalmers E, Brown S, Keeling D, et al. Early factor VIII exposure and subsequent inhibitor development in children with severe haemophilia A. Haemophilia. 2007;13:149–155.
- Eckhardt C, Van der Bom J, van der Naald M, et al. Surgery and inhibitor development in hemophilia A: a systematic review. J Thromb Haemost. 2011;9:1948–1958.