
ABSTRACT
Introduction: During the past few years, new genomic approaches have elucidated the molecular genetics of chronic lymphocytic leukemia (CLL) to a large extent. As a consequence, specific high-risk genetic features of the disease, e.g. TP53 disruption, have become the backbone of the treatment algorithm for CLL and serve as robust biomarkers for a precision medicine approach to this leukemia.
Areas covered: This review covers the genetics of CLL and highlights the translational implications of molecular biomarkers that characterize patients with a high risk of disease progression. Knowledge of the genetic landscape of CLL has allowed the identification of the main molecular features associated with chemo-refractoriness, as well as resistance to BCR inhibitors and BCL2 inhibitors. The molecular basis of Richter transformation has also been characterized.
Expert opinion: The term ‘high risk CLL’ has been changing over time, and might be subject to further changes in the future. With the advent of new therapeutic strategies targeting pathogenetic pathways of the disease, the definition is shifting from the historical view of refractoriness to chemo-immunotherapy, to refractoriness to BCR inhibitors and/or to BCL2 inhibitors. Patients failing these novel medicines are those for whom new therapeutic approaches are still highly needed.
1. Introduction
Chronic lymphocytic leukemia (CLL) is characterized by a high degree of molecular heterogeneity that reflects the highly variable clinical behavior of the disease [Citation1]. In the past decade, a large body of genomic investigations have deciphered the genome of CLL, at least to a significant extent. Elucidation of the CLL genomic landscape has confirmed the notion that CLL is not associated with a unique genetic lesion but, conversely, that this leukemia harbors many different genetic abnormalities that may interact and/or surrogate and complement in inducing the development and progression of CLL () [Citation2,Citation3]. These molecular findings have also been translated into the clinical practice, helping clinicians to offer a more precise treatment for every single patient based on the mutational profile of the disease [Citation1]. More precisely, according to the 2018 iwCLL guidelines [Citation4], the following biomarkers should be tested before treatment start: i) TP53 disruption, including both TP53 mutation and 17p deletion, ii) 11q deletion, 13q deletion and trisomy 12, and iii) immunoglobulin heavy chain variable (IGHV) gene mutational status. Moreover, complex karyotype, defined by the presence of at least 3 chromosome aberrations by cytogenetic analysis, has been associated with shorter survival in CLL patients, though this test has not entered routine clinical practice [Citation4]. On these grounds, CLL has become a model for the application of Precision Medicine in the field of hematological neoplasms.
Figure 1. Genetic lesions involved in CLL clonal evolution and in Richter transformation. CLL is characterized by a high degree of molecular heterogeneity. During the clinical history of the disease, CLL may acquire new genetic lesions that may predispose to treatment resistance and, eventually, to Richter transformation. According to this model, at the time of diagnosis the CLL clone is characterized by a preponderance of cells that are sensible to chemotherapy (in yellow), and by only few cells that harbor genetic lesions, such as TP53 abnormalities, predisposing to chemo-refractoriness (in red). At the time of refractoriness to CIT, the CLL clone is mainly composed by cells (in red) that were not cleared by the previous chemotherapy because they harbor genetic lesions that confer refractoriness to CIT. Subsequent lines of treatment include BCRi (ibrutinib targeting BTK and idelalisib targeting PI3Kδ) and the BCL2i venetoclax. Despite the high efficacy of these drugs, a fraction of patients fails to respond over time. The mechanisms of refractoriness to ibrutinib and to venetoclax have been identified, at least to a certain extent. In particular, mutations of the BTK binding domain of ibrutinib, as exemplified in the cells colored in blue, and mutations of the BCL2 binding site of venetoclax, as exemplified in the cells colored in orange, may cause resistance to ibrutinib and to venetoclax, respectively. In addition, at every time point of the CLL clinical history, specific genetic abnormalities (in purple) involving the c-MYC, TP53, NOTCH1 and CDKN2A genes may predispose or lead to Richter transformation, represented by the histologic evolution of CLL to an aggressive B-cell lymphoma.
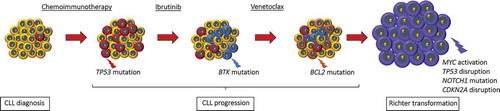
The most consolidated example of Precision Medicine in the treatment algorithm of CLL is represented by the occurrence of TP53 abnormalities, including both mutations and deletions, that identify a group of very high-risk CLL patients that are destined to refractoriness to chemo-immunotherapy (CIT), independent of the chemotherapeutic backbone that is utilized. Consistently, these patients are now treated in first line with B-cell receptor inhibitors (BCRi), namely ibrutinib, or with BCL2 inhibitors (BCL2i), namely venetoclax, that can, at least in part, circumvent the chemorefractoriness associated with TP53 disruption () [Citation4]. Another example of the application of Precision Medicine in CLL treatment is represented by the mutation status of the IGHV genes, since it affects responsiveness and long term outcome to CIT [Citation5–Citation7].
Figure 2. Molecular pathways targeted by ibrutinib and venetoclax and mechanisms of resistance to these drugs. Ibrutinib is a BTK inhibitor that acts by blocking the BTK catalytic site. The inactivation of the BCR pathway mediated by ibrutinib leads to the inhibition of the NF-κB pathway, thus reducing CLL cell proliferation and promoting apoptosis. The mode of action of ibrutinib is independent of TP53 disruption that represents the most robust predictor of chemo-refractoriness. Ibrutinib treatment may lead to the emergence of BTK missense mutations targeting codon 481 (p.C481S, leading to the cysteine-to-serine amino acid change) and thus altering the binding of the drug to BTK and causing the loss of its therapeutic effect. Venetoclax is a potent and selective BH3-mimetic drug that binds and blocks the BCL2 anti-apoptotic protein, leading to apoptosis that is independent of DNA damage response (and independent of disruption of TP53 that modulates the DNA damage response). A single nucleotide variant in the BCL2 gene (c.302G>T, p.Gly101Val) targets the BCL2 protein binding site of the drug, and may account for approximately 50% of cases of venetoclax resistance.
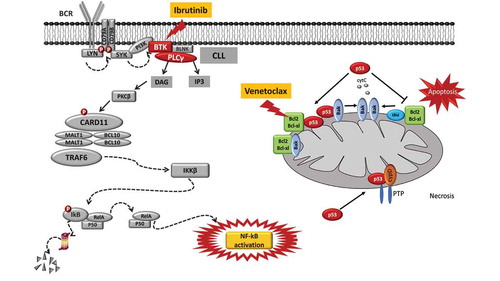
Until the introduction of BCRi and BCL2i, namely venetoclax, CLL was defined as high risk if it displayed the following characteristics: i) refractoriness to purine analogs; ii) relapse within two years after CIT; and iii) presence of deletion and/or mutation of the TP53 gene [Citation8]. However, as recently proposed, after the introduction of biological medicines, the definition of high risk CLL may be changed to include two new categories: i) patients with TP53 abnormalities who have failed CIT but respond either to a BCRi or to a BCL2i; ii) patients who, independent of TP53 status, have failed both CIT and a first BTKi or BCL2i; and iii) patients who progressed after receiving a novel targeted agent, e.g. BCRi or BCL2i [Citation8].
In this review, we shall provide a brief coverage of the genetic landscape of CLL, and we will focus in detail on the genetics of high risk patients who fail CIT and/or the novel biological drugs. We then briefly describe the genetic lesions that associate with Richter transformation, a very high risk evolution of CLL into an aggressive lymphoma.
2. Genetic landscape of CLL
Whole exome sequencing (WES) and whole genome sequencing (WGS) studies have enabled a comprehensive identification of the cancer-associated genes recurrently involved in CLL. The CLL genome is largely devoid of chromosomal translocations and of aberrant somatic hypermutation that are otherwise involved in several types of B cell non-Hodgkin lymphomas (B-NHLs) [Citation9]. Considering clinically unselected CLL cases, the typical genome carries ~2000 molecular lesions, among which, however, only few mutations recur across patients at a frequency higher than 5%, whereas a large number of biologically and clinically uncharacterized genes are mutated at lower frequencies [Citation2,Citation3].
The genome of CLL, similarly to other tumors, is dynamic and the genetic lesions may change over time, leading to clonal evolution of the disease and to the emergence of more aggressive clones or subclones. A paradigmatic example of clonal evolution in CLL is represented by the accumulation of TP53 alterations or BIRC3 mutations, whose frequency is consistently higher at the time of chemo-refractoriness than at the time of diagnosis [Citation10,Citation11]. Despite the numerous novel insights into the molecular genetics of CLL, the detailed mechanisms responsible for disease initiation and for the diversity in clonal evolution across patients remain largely unknown.
As stated above, CLL does not harbor a single and unifying genetic alteration that associates with all cases. Rather, a variety of molecular pathways are involved in the disease. Among the most recurrent genetic events, deletion of chromosome13q14 (del13q14) has been considered among the early drivers of CLL and is overall the most frequent genetic abnormality in this leukemia, being present in more than 50% of CLL cases [Citation2,Citation3]. Del13q14 causes the loss of two microRNA (miRNA), namely miR-15a and miR-16 [Citation12]. As a consequence, loss of miR-15a and miR-16 abrogates or reduces the negative control of BCL2 translation, leads to enhanced levels of BCL2 protein expression, and thus contributes to enhanced cell survival of CLL cells [Citation13].
Recurrent mutations are not homogeneously spread across the CLL genome, but, rather, affect genes that can be integrated into a limited set of pathways. These include microenvironment-dependent signaling pathways through NOTCH (NOTCH1, FBXW7), inflammatory receptors (MYD88), MAPK–extracellular signal-regulated kinase (BRAF, KRAS, NRAS, MAP2K1) and NF-kB pathways (BIRC3, TRAF3, NFKBIE), as well as intracellular programs such as DNA damage and cell cycle control (ATM, TP53, SAMHD1, POT1), chromatin modification (HIST1H1E, CHD2, ZMYM3), transcription (EGR2, IRF4, BCOR, MED12), and ribosomal processing (XPO1, SF3B1, RPS15) [Citation1].
3. Molecular abnormalities of patients failing CIT
Chemotherapy agents exert their antineoplastic activity by interfering with the normal structure of cellular genomic DNA. In normal cells, if the DNA damage induced by chemotherapy cannot be restored by the physiological DNA repairing enzymes, a cascade of intracellular signaling (namely, related to TP53 and ATM functions) causes the apoptosis of the cell. On these grounds, the correct function of the proteins involved in the DNA damage response machinery is essential for inducing cell death in cells treated with chemotherapy. Among these proteins, TP53 plays a pivotal role in the DNA repair pathway. Loss of TP53 function, by mutation and/or deletion, can prevent cell apoptosis, thus leading to the survival and subsequent proliferation of cells with an increased degree of complexity in the number and type of chromosomal and genetic abnormalities [Citation14,Citation15].
The CIT regimen containing fludarabine, cyclophosphamide and rituximab (FCR) has represented a breakthrough in the management of young and fit CLL patients with an improvement in both PFS and OS compared to previous regiments [Citation16]. However, both in clinical trials and in real life cohorts, TP53 disruption, including both 17p deletion and TP53 mutations, sorted out as a robust predictor of poor response to FCR [Citation5–Citation7,Citation17]. The association between TP53 disruption and CIT failure is not limited to the case of FCR, but has been clearly documented also for other CIT regimens, namely bendamustine-rituximab and obinutuzumab-chlorambucil [Citation18,Citation19]. Consistently, all guidelines converge in stating that TP53 disrupted patients in the clinical practice should not be treated with CIT, but with alternative therapeutic strategies that can circumvent, at least in part, the TP53-associated chemo-refractoriness [Citation4]. Despite the important role of TP53 disruption as a predictor of CIT failure, this molecular biomarker does not fully capture all high-risk patients destined to relapse after CIT, and accounts for only 30%-40% of CLL refractory to fludarabine [Citation5–Citation7,Citation17].
Several molecular studies have identified a set of genes, including NOTCH1, SF3B1 BIRC3, RSP15, and EGR2, as novel molecular biomarkers that account, at least in part, for chemo-refractoriness in addition to TP53 disruption. These genetic aberrations appear to be mutually exclusive with disruption of TP53, at least in most cases, indicating that multiple alternative pathways can lead to CIT failure [Citation11,Citation20–Citation23]. In particular, in the case of FCR, mutations of BIRC3 (Baculoviral IAP Repeat Containing 3) stand out as a novel predictor of treatment failure. Notably, BIRC3 mutations are rare at the time of CLL diagnosis (3–4%), but are detectable in approximately 25% of fludarabine refractory patients [Citation11]. In a multicenter cohort of CLL treated with FCR, next generation sequencing analysis of a set of genes recurrently mutated in CLL revealed that BIRC3 mutations identify a poor prognostic subgroup of patients failing FCR similar to cases harboring TP53 disruption [Citation24]. BIRC3 mutations maintained an independent association with an increased risk of progression in multivariate analysis adjusted for TP53 mutation, 17p deletion and mutation status of IGHV genes [Citation24]. Interestingly, mutations of BIRC3 have also been reported to predict poor outcome in the obinutuzumab-chlorambucil arm of the CLL-14 trial, comparing front line obinutuzumab-chlorambucil versus obinutuzumab-venetoclax [Citation19]. BIRC3 ubiquitinates and negatively regulates components of the non-canonical NF-κB pathway, namely MAP3K14 (also known as NIK), a positive regulator of cell proliferation and survival [Citation25,Citation26]. Consistent with this function in normal physiology, BIRC3 mutations associate with activation of the non-canonical NF-κB pathway [Citation24].
4. Molecular abnormalities of patients failing BCRi
Ibrutinib is an oral BTK inhibitors approved for first line treatment of CLL patients with TP53 abnormalities and of patients unsuitable for CIT, as well as for subsequent lines of therapy in relapsed/refractory cases.
In CLL patients carrying TP53 abnormalities, clinical trials have demonstrated a high efficacy of ibrutinib and idelalisib in the context of both high risk relapsed/refractory patients and in treatment naïve patients [Citation27–Citation29]. The recent update of two clinical trials with a 5-year follow-up of single-agent ibrutinib has highlighted the high efficacy of the drug and the long lasting durability of the response to ibrutinib, and also has allowed to stratify the outcome of ibrutinib treated patients according to biomarkers, namely TP53 disruption and complex karyotype [Citation30,Citation31]. In the trial by Ahn et al., in which most patients were treatment naïve, TP53 disruption, together with a prior history of treatment, was identified as an important determinant of progression-free survival (PFS) on ibrutinib [Citation30]. It may well be that TP53 disruption had been accumulated and positively selected by previous lines of ineffective CIT. Whether ibrutinib itself may positively select for TP53 disruption in CLL is still a matter of investigation.
Similar to the trial by Ahn et al., also in the trial by O’Brien et al., ibrutinib yielded a high overall response rate and a PFS that had never been previously achieved with other drugs in the context of relapsed/refractory CLL [Citation31]. Also in this trial, the best efficacy of ibrutinib was obtained in TP53 wild type patients as well as in treatment naïve patients. Remarkably, complex karyotype, defined as the occurrence of ≥3 chromosomal abnormalities, sorted out as a predictor of poor response of ibrutinib [Citation31]. The unfavorable impact of complex karyotype in CLL treated with ibrutinib has been documented in other studies where complex karyotype was independently associated with disease progression and inferior survival [Citation32]. However, when considering different trials, the prognostic value of complex karyotype has not been identified as an independent factor in all studies, and in some series it appears to be largely influenced by the co-existence of del17p [Citation33]. Therefore, the true significance of complex karyotype per se as a biomarker of BCRi failure is still under scrutiny, and complex karyotype is not consistently considered as a biomarker of high risk CLL.
In the context of CIT, the mutational status of IGHV genes correlates with disease outcome. In particular, the occurrence of unmutated IGHV genes (e.g. ≥98% of identity to the germline counterpart) predicts poor outcome to several CIT regimens [Citation4,Citation5,Citation18]. Conversely, CLL carrying mutated IGHV genes (e.g. <98% of identity to the germline counterpart), in the absence of TP53 disruption, may have a durable response to CIT [Citation4–Citation6]. At variance with the predictive value in patients treated with CIT, the mutation status of IGHV genes appears to have no significant relevance in cases treated with BCRi, since these drugs are able to overcome the poor prognosis associated with unmutated IGHV genes [Citation30,Citation31].
In addition to TP53 disruption and, potentially, to complex karyotype, other genetic alterations have been proposed as biomarkers of reduced efficacy of BCRi. The Resonate clinical trial is a phase 3 study designed to compare the efficacy and safety of ibrutinib versus ofatumumab in relapsed/refractory CLL patients [Citation27]. The long term follow up of the Resonate study showed that, in addition to TP53 disruption, also mutations of SF3B1, a component of the spliceosome machinery, showed a trend toward and inferior PFS in the ibrutinib arm [Citation34].
TP53 disruption and potentially complex karyotype lead to reduced efficacy of ibrutinib with a molecular mechanism that is independent of the site of action and of the precise mode of action of ibrutinib. In fact, ibrutinib exerts its function by binding to a cysteine residue positioned at codon 481 of the BTK gene, thus blocking the catalytic site of the Bruton tyrosine kinase. A pivotal whole exome sequencing analysis has documented the existence of specific mechanisms of resistance to ibrutinib due to mutations [Citation35]. Ibrutinib treatment may lead to the emergence of BTK missense mutations targeting codon 481 (p.C481S, leading to a cysteine-to-serine amino acid change), thus altering the binding of the drug to BTK and causing the loss of ibrutinib therapeutic effect [Citation35]. These mutations are absent in ibrutinib-naïve CLL, and may be selected upon exposure to the drug [Citation36]. Functional analysis has shown that the C481S mutation of BTK results in a protein that prevents the irreversible covalent binding of ibrutinb to its target [Citation35,Citation37]. An alternative mechanism of resistance to ibrutinib is represented by the constitutive activation of proteins located downstream to BTK. Because ibrutinib acts upstream of such proteins, their signaling is no longer affected by ibrutinib inhibition once that such components are constitutively activated by mutations. Consistent with this model, several mutations in the PLCγ2 gene (R665W and L845F are the most frequent mutations) detected in ibrutinib-resistant CLL are potentially gain-of-function mutations that lead to autonomous B-cell receptor activity [Citation35,Citation37,Citation38]. Overall, acquired mutations of BTK or PLCγ2 have been detected in approximately 85% patients failing ibrutinib treatment [Citation35–Citation38]. Consistently, patients with BTK or PLCγ2 mutations may be considered as high risk CLL patients for whom subsequent lines of therapy with innovative drugs are required. Using high sensitivity techniques, the emergence of BTK or PLCγ2 mutations has been shown to antedate clinical relapse by a median of 9.3 months before [Citation35–Citation38]. However, regular monitoring of BTK or PLCγ2 mutations is not recommended by current guidelines [Citation4].
5. Molecular abnormalities of patients failing BCL2i
Venetoclax, a small molecule representing a second generation BCL2i, has a significant impact on the natural history of CLL both in relapsed/refractory patients and in TP53 disrupted cases [Citation39,Citation40]. The high efficacy of venetoclax also in these high-risk groups of patients may rely on the fact that the apoptotic pathway induced by the drug is largely independent of the DNA repair pathway that is frequently altered in CLL and contributes to refractoriness to chemotherapeutic agents [Citation41]. Normal murine nodal B cells, human B-lymphoblast cell lines, and primary CLL cells carrying TP53 disruption maintain sensitivity in vitro to apoptosis induction by venetoclax, in a fashion similar to cells with a normal TP53 function [Citation41]. These pre-clinical findings may explain the high efficacy of venetoclax in high-risk CLL patients.
Despite the short follow up and the limited number of progressions upon venetoclax treatment, a few studies have tried to reveal the genetical lesions that characterize high risk patients destined to relapse upon venetoclax treatment [Citation42–Citation44]. In one of the first molecular analysis of patients treated with venetoclax, eight patients were analyzed by whole-exome-sequencing before the initiation of venetoclax therapy and at the time of venetoclax resistance [Citation42]. All patients had shown a significant clinical response to venetoclax before developing disease progression or relapse. In most patients, mutations in cancer-related genes (i.e. BRAF, CD274, NOTCH1, RB1, SF3B1, and TP53) were identified. The variant allele frequency of such mutations dynamically changed during venetoclax treatment and at the time of progression [Citation42]. More precisely, this study showed that molecular lesions associated with venetoclax resistance may include: i) recurrent mutations in the BTG1 gene; ii) homozygous deletions affecting CDKN2A/B that developed during treatment; iii) mutations in the BRAF gene; and iv) a high-level focal amplification of CD274 (PD-L1) [Citation42]. Notably, BRAF mutations and CD274 amplifications might be used for targeted therapies with BRAF inhibitors or immune-checkpoint inhibitors, respectively [Citation42].
In a second study, sixty-seven consecutive patients with relapsed/refractory CLL treated with venetoclax were retrospectively analyzed [Citation43]. After a median follow-up of 23 months, 25 (37%) patients had developed disease progression: 14 with Richter transformation (RT) to diffuse large B cell lymphoma (DLBCL), 3 with RT to Hodgkin lymphoma (HL), and 8 with progressive CLL. In this study, fludarabine-refractoriness and complex karyotype were the two dominant risk factors for progression despite ongoing treatment with the BCL2 inhibitor [Citation43]. Of interest, these findings suggest that complex karyotype may hold greater prognostic significance than TP53 aberrations in heavily pretreated patients receiving novel agents, possibly reflecting greater genomic instability [Citation43].
Despite the potential relevance of alterations of BRAF, CD274, NOTCH1, RB1, SF3B1, and TP53 and of complex karyotype in determing venetoclax resistance, none of these molecular lesions directly affect the mode of action of venetoclax. Similar to the molecular model of ibrutinib resistance, also in the case of venetoclax mutations in the protein binding domain of the drug have been disclosed [Citation44]. Pre- and post-progression samples of CLL patients treated with venetoclax in three early phase clinical trials have been analyzed by next generation sequencing. A single heterozygous nucleotide variant was detected in BCL2, namely c.302G>T, p.Gly101Val, in 7 out of 15 patients who progressed during venetoclax [Citation44]. Further investigations using a highly sensitive and specific droplet digital PCR (ddPCR) assay indicated that the Gly101Val mutation was already detectable at low variant allele fraction up to 25 months before the standard disease progression criteria were met. The Gly101Val mutation was not detected prior to venetoclax treatment in this cohort [Citation44]. Functional studies have demonstrated that cells that have been modified to express the Gly101Val mutation in BCL2 are 30-fold less sensitive to venetoclax than cells expressing wild-type BCL2. Venetoclax, in fact, is a BIM-like molecule that binds to BCL2 with a greater affinity than the pro-apoptotic protein BIM. In binding assays, the capacity for venetoclax to compete in vitro with BIM for binding to the Gly101Val mutant was markedly reduced (180-fold) compared to wild-type BCL2. Regarding the physiological activity of BCL2, in the absence of venetoclax the Gly101Val mutant demonstrated preserved normal function by protecting cell lines from apoptosis induced by cytotoxic drugs with similar effectiveness to wild-type BCL2 [Citation44].
6. Molecular features of richter transformation
RT is defined as the occurrence of an aggressive B-cell lymphoma in patients with a previous or concomitant diagnosis of CLL. Two pathologic variants of RT are currently recognized, namely the DLBCL variant and the HL variant [Citation45]. After the diagnosis of RT, it is important to evaluate the clonal relationship of the RT clone with the preexistent CLL clone. The assessment of the clonal relationship between CLL and RT allows to identify two different groups of patients with a different risk of progression and death. Patients with clonally related RT, namely patients in which the preexistent CLL phase and the RT phase carry the same IGHV gene rearrangement, represent a very high risk group of patients [Citation46–Citation48]. Consistently, because of their poor outcome, clonally related RT, if age and fitness allow, are usually consolidated with allogeneic stem cell transplantation after induction treatment with CIT. On the other hand, clonally unrelated RT represent a different molecular and clinical subgroup of RT patients that are characterized by a better outcome and may benefit from R-CHOP without further treatment [Citation46–Citation48].
The molecular profile of RT with DLBCL features is heterogeneous, lacks a unifying genetic lesion, and does not fully overlap with the genetic landscape of de novo DLBCL [Citation46]. Genetic lesions of RT with DLBCL features recurrently involve the TP53, NOTCH1, MYC, and CDKN2A genes, that regulate apoptosis and proliferation and are usually associated to a chemo-refractory phenotype [Citation49–Citation51]. More precisely, TP53 is mutated in more than 60% of DLBCL RT. TP53 mutations may be acquired at the time of transformation if they are not already present in the CLL phase. TP53, as discussed earlier, is a major player of the DNA damage response, and its disruption in RT may explain the chemo-refractory phenotype that is consistently shown by DLBCL-type RT [Citation49,Citation51]. The CDKN2A gene codes for a tumor suppressor protein that negatively regulates the cell cycle. Its inactivation by deletion explains, at least in part, the high proliferation rate of RT [Citation49]. CDKN2A deletions are found in 30% of cases of RT, and are generally acquired at the time of transformation [Citation50]. The MYC network is deregulated in 70% of DLBCL-type RT by structural alterations of the c-MYC gene, represented by chromosomal translocations and amplification. At variance with de novo DLBCL, the presence of c-MYC abnormalities in RT does not display concurrent BCL2 or BCL6 translocations. In the remaining cases, the c-MYC pathway may be also deregulated, by truncating mutations and deletions of the c-MYC negative regulator MGA and by mutations activating MYC trans-regulatory factors, such as NOTCH1 [Citation51–Citation53]. Beside the occurrence of molecular alterations of the tumor clone, also the immunogenotype of the clone is important for RT development. Notably, the usage of specific stereotyped immunoglobulin genes in the subset 8 configuration (IGHV4-39/IGHD6-13/IGHJ5) is highly enriched in DLBCL RT, supporting a role of B-cell receptor signaling in transformation [Citation54].
Molecular lesions that are frequently present in de novo DLBCL, such as inactivating mutations of the acetyltransferase genes CREBBP/EP300 and of the B2M gene, coding for a protein associated with the major histocompatibility complex (MHC) class I, as well as common translocations involving BCL6 and/or BCL2, are usually absent in RT DLBCL pateints [Citation46]. These differences indicate that DLBCL transformed from CLL and de novo DLBCL represent distinct disease entities.
The genetics of RT has been investigated predominantly in the era of CIT treatment. During the last few years, an increasing number of CLL patients have been treated with biological agents, namely BCRi or venetoclax, either as first line therapy or in the relapsed/refractory setting. The molecular mechanisms leading to RT in the era of new CLL drugs are not completely understood. In the few available studies, complex karyotype, TP53 disruption and 8q24 abnormalities have been suggested to be associated with RS [Citation55]. Although BTK or PLCG2 mutations predispose to ibrutinib resistance and are known to expand at the time of CLL progression, mutations of these genes are rarely found in RT that emerge after ibrutinib treatment [Citation33,Citation55].
7. Conclusions
The advent of novel agents is having a profound impact on both the clinical history of CLL and on the molecular and clinical features defining high-risk disease. Traditionally, TP53 alterations have been considered the most important genetic lesion associated with refractoriness to CIT. However, with the advent of novel biological drugs that are able to overcome TP53-related chemorefractoriness, the role of TP53 disruption in defining high risk CLL is evolving. In fact, BCRi (ibrutinib and idelalisib) and BCL2i (venetoclax) exert their function in a TP53 independent manner, and their action is not substantially affected by TP53 disruption. However, it should be noted that BCRi or BCL2i mitigate the detrimental effect of TP53 disruption, but do not abolish it completely. This finding highlights the importance of a better understanding of the TP53 pathway in relationship with the effects of BCRi and BCL2i.
On the one hand, the introduction of BCRi and BCL2i has overcome, at least in part, the unfavorable impact of aggressive genetic lesions in the context of CIT. On the other hand, however, the advent of these new drugs has been accompanied by the onset of new resistance mutations. Regarding ibrutinib, BTK and PLCγ2 mutations pose a concern to the long-term efficacy of ibrutinib therapy. Consistently, novel non-covalent BTK inhibitors are under investigation with the aim of overcoming these detrimental genetic lesions [Citation56]. Similarly, BCL2 mutations have been recently described and pose new challenges in the setting of high risk CLL patients who progress after venetoclex therapy
8. Expert opinion
The term ‘high risk CLL’ has been changing over time. Until five to ten years ago, the definition of high risk CLL included patients who were refractory to CIT or the ones who harbored TP53 disruption, which represents the strongest predictor of chemo-refractoriness. With the advent of new drugs, namely BCRi and BCL2i, the scenario of high risk CLL has been changing since these drugs have demonstrated a high efficacy in both chemo-refractory patients and in TP53 disrupted patients. The high efficacy of these biological drugs relies on the fact that they: i) act in a TP53 independent manner; and ii) target two of the most important pathogenetic pathways in CLL, namely BCR signaling and apoptosis. However, treatment with BCRi and BCL2i may face the emergence of resistant clones that can reduce the efficacy of these novel drugs. The main mechanisms of resistance to novel drugs are directly related to the drug mode of action, and imply mutations of BTK and PLCγ2 in the case of ibrutinib resistance, and mutations of BCL2 in the case of venetoclax refractoriness. These mutations are absent before treatment and emerge during the treatment course predisposing to the loss of function of the drug. However, these mutations do not involve the entire CLL clone, and further studies are required to demonstrate whether these acquired genetic lesions are sufficient per se to cause refractoriness, or whether other genetic alterations are needed for driving CLL progression. The development of novel strategies that might overcome these new types of refractoriness are needed to improve patient outcome in these high risk settings.
In recent years, minimal residual disease (MRD) has emerged as a strong predictor of outcome both in patients treated with CIT and in patients treated with biological drugs. In patients treated with BCRi or BCL2i, MRD evaluation may become a potential tool to decide the timing of drug interruption. To date, outside of clinical trials, BCRi or BCL2i are administered until progression without a fixed duration schedule, in the absence of major toxicities or intolerance. However, recent evidence suggests that patients treated with combinations of anti-CD20 antibodies and venetoclax who achieve a deep MRD negativity may remain free from progression for a longer time compared to patients with higher levels of MRD [Citation57,Citation58]. Consistently, several different clinical trials are now enrolling patients with the aim of identifying the best time for treatment discontinuation based on both MRD monitoring and on other biological features of the disease.
The outcome of CLL patients has dramatically improved during the last decades thanks to the introduction of more effective CIT regiments and to the introduction of biological drugs, namely BCRi and BCL2i. Moreover, the large body of genomic investigations have deciphered the genome of CLL and have identified molecular predictors, namely TP53 disruption and IGHV mutations status, that allow clinicians to select the more effective therapeutic agent or drug combination for every single patient. MRD monitoring may further help in tailoring treatment duration. Many ongoing clinical trials in the first line setting are now testing different combinations of drugs with the aim of identifying the best combination that can potentially eradicate the CLL clone with a fixed duration schedule.
Article Highlight Box
The marked degree of molecular heterogeneity of CLL reflects the clinical heterogeneity of the disease.
High-risk genetic features of the disease, namely TP53 disruption and unmutated IGHV genes, are the backbone of the CLL treatment algorithm, and serve as robust biomarkers for a precision medicine approach to this leukemia.
The term ‘high risk CLL’ has changed over time. Until the introduction of BCRi and BCL2i, the following features defined high risk CLL: i) refractoriness to purine analogs; ii) relapse within two years after CIT; and iii) presence of deletion and/or mutation of the TP53 gene.
After the advent of BCRi and BCL2i, the definition of high risk CLL now encompasses: i) patients with TP53 abnormalities who have failed CIT but respond either to a BCRi or to a BCL2i; ii) patients who, independent of TP53 status, have failed both CIT and a first BTKi or BCL2i; and iii) patients who have progressed after receiving a novel targeted agent, e.g. BCRi or BCL2i.
CLL outcome has drastically improved during recent times thanks to the introduction of inhibitors of the BCR and of BCL2 and of novel anti-CD20 MoAbs. Many ongoing clinical trials are now testing different combinations of drugs in different molecular subtypes of the disease with the aim of potentially eradicating the CLL clone.
Declaration of interest
G Gaidano has received the following research grants from academic institutions: Molecular bases of disease dissemination in lymphoid malignancies to optimize curative therapeutic strategies, (5 x 1000 No. 21,198), Associazione Italiana per la Ricerca sul Cancro Foundation Milan, Italy; Progetti di Rilevante Interesse Nazionale (PRIN), (2015ZMRFEA), Rome, Italy; the AGING Project – Department of Excellence – DIMET, Università del Piemonte Orientale, Novara, Italy; and Ricerca Finalizzata 2018 (project RF-2018-12,365,790), MoH, Rome, Italy. He has also discloses roles in advisory boards or speakers’ bureaus of the following companies: Astra-Zeneca, Sunesys, Abbvie and Janssen. A Patriarca is a consultant of Novartis and Novonordisk. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Gaidano G, Rossi D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematology Am Soc Hematol Educ Program. 2017;2017(1):329–337.
- Landau DA, Tausch E, Taylor-Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525–530.
- Puente XS, Beà S, Valdés-Mas R, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–524.
- Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760.
- Rossi D, Terzi-di-Bergamo L, De Paoli L, et al. Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood. 2015;126(16):1921–1924.
- Thompson PA, Tam CS, O’Brien SM, et al. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood. 2016;127(3):303–309.
- Fischer K, Bahlo J, Fink AM, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208–215.
- Dreger P, Ghia P, Schetelig J, et al. High-risk chronic lymphocytic leukemia in the era of pathway inhibitors: integrating molecular and cellular therapies. Blood. 2018;132(9):892–902.
- Gaidano G, Foà R, Dalla-Favera R. Molecular pathogenesis of chronic lymphocytic leukemia. J Clin Invest. 2012;122(10):3432–3438.
- Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123(14):2139–2147.
- Rossi D, Fangazio M, Rasi S, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood. 2012;119(12):2854–2862.
- Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99(24):15524–15529.
- Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci. 2005;102:13944–13949.
- Hientz K, Mohr A, Bhakta-Guha D, et al. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget. 2017;8(5):8921–8946.
- Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28(3):128–136.
- Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174.
- Stilgenbauer S, Schnaiter A, Paschka P, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123(21):3247–3254.
- Eichhorst B, Fink AM, Bahlo J, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17(7):928–942.
- Tausch E, Bahlo J, Robrecht S, et al. Genetic markers and outcome in the CLL14 trial of the GCLLSG comparing front line obinutuzumab plus chlorambucil or venetoclax in patients with comorbidity. HemaSphere. 2019;3:4.
- Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119(2):521–529.
- Rossi D, Bruscaggin A, Spina V, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118(26):6904–6908.
- Ljungström V, Cortese D, Young E, et al. Whole-exome sequencing in relapsing chronic lymphocytic leukemia: clinical impact of recurrent RPS15 mutations. Blood. 2016;127(8):1007–1016.
- Young E, Noerenberg D, Mansouri L, et al. EGR2 mutations define a new clinically aggressive subgroup of chronic lymphocytic leukemia. Leukemia. 2017;31(7):1547–1554.
- Diop F, Moia R, Favini C, et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica. 2019. [Epub ahead of print]. DOI:10.3324/haematol.2019.219550
- Vince JE, Wong WW, Khan N, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131(4):682–693.
- Jost PJ, Ruland J. Aberrant NF-kappaB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood. 2007;109(7):2700–2707.
- Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–223.
- Sharman JP, Coutre SE, Furman RR, et al. Final results of a randomized, phase III study of rituximab with or without idelalisib followed by open-label idelalisib in patients with relapsed chronic lymphocytic leukemia. J Clin Oncol. 2019;37(16):1391–1402.
- Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–2437.
- Ahn IE, Farooqui MZH, Tian X, et al. Depth and durability of response to ibrutinib in CLL: 5-year follow-up of a phase 2 study. Blood. 2018;131(21):2357–2366.
- O’Brien S, Furman RR, Coutre S, et al. Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood. 2018;131(17):1910–1919.
- Thompson PA, O’Brien SM, Wierda WG, et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer. 2015;121(20):3612–3621.
- Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1(1):80–87.
- Byrd JC, Hillmen P, O’Brien S, et al. Long-term follow-up of the RESONATE™ phase 3 trial of ibrutinib versus ofatumumab. Blood. 2019;133:2031–2042. [Epub ahead of print].
- Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–9224.
- Famà R, Bomben R, Rasi S, et al. Ibrutinib-naïve chronic lymphocytic leukemia lacks Bruton tyrosine kinase mutations associated with treatment resistance. Blood. 2014;124(25):3831–3833.
- Woyach JA, Ruppert AS, Guinn D, et al. BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35(13):1437–1443.
- Burger JA, Landau DA, Taylor-Weiner A, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016;20(7):11589.
- Moia R, Diop F, Favini C, et al. Potential of BCL2 as a target for chronic lymphocytic leukemia treatment. Expert Rev Hematol. 2018;11(5):391–402.
- Gentile M, Petrungaro A, Uccello G, et al. Venetoclax for the treatment of chronic lymphocytic leukemia. Expert Opin Investig Drugs. 2017;26(11):1307–1316.
- Anderson MA, Deng J, Seymour JF, et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood. 2016;127:3215–3224.
- Herling CD, Abedpour N, Weiss J, et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat Commun. 2018;9(1):727.
- Anderson MA, Tam C, Lew TE, et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood. 2017;129(25):3362–3370.
- Blombery P, Anderson MA, Gong JN, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019;9(3):342–353.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390.
- Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131(25):2761–2772.
- Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12):3391–3401.
- Parikh SA, Kay NE, Shanafelt TD. How we treat Richter syndrome. Blood. 2014;123(11):1647–1657.
- Fabbri G, Khiabanian H, Holmes AB, et al. Genetic lesions associated with chronic lym- phocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210(11):2273–2288.
- Chigrinova E, Rinaldi A, Kwee I, et al. Two main genetic pathways lead to the trans- formation of chronic lymphocytic leukemia to Richter syndrome. Blood. 2013;122(15):2673–2682.
- Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding ge- nome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389–1401.
- Fabbri G, Holmes AB, Viganotti M, et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2017;114(14):E2911–E2919.
- De Paoli L, Cerri M, Monti S, et al. MGA, a suppressor of MYC, is recurrently inactivated in high risk chronic lymphocytic leukemia. Leuk Lymphoma. 2013;54(5):1087–1090.
- Rossi D, Spina V, Cerri M, et al. Stereotyped B-cell receptor is an independent risk factor of chronic lymphocytic leukemia trans- formation to Richter syndrome. Clin Cancer Res. 2009;15(13):4415–4422.
- Kadri S, Lee J, Fitzpatrick C, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017;1(12):715–727.
- Allan JN, Wierda WG, Patel K, et al. Preliminary safety, pharmacokinetic, and pharmacodynamic results from a phase 1b/2 dose-escalation and cohort-expansion study of the noncovalent, reversible Bruton’s Tyrosine Kinase Inhibitor (BTKi), vecabrutinib, in B-lymphoid malignancies. Blood. 2018;132:3141.
- Kater AP, Seymour JF, Hillmen P, et al. Fixed duration of venetoclax-rituximab in relapsed/refractory chronic lymphocytic leukemia eradicates minimal residual disease and prolongs survival: post-treatment follow-up of the MURANO phase III study. J Clin Oncol. 2019;37(4):269–277.
- Fischer K, Ritgen M, Al-Sawaf O, et al. Quantitative analysis of minimal residual disease (MRD) shows high rates of undetectable MRD after fixed-duration chemotherapy-free treatment and serves as surrogate marker for progression-free survival: a prospective analysis of the randomized CLL14 trial. ASH. 2019;134(Supplement_1):36.