
KEYWORDS:
1. Introduction
We are molecular toolmakers. The tools we design, synthesize, and create disrupt or repair the human genome. Either outcome can be achieved using bio-molecular scissors generally classified as programmable nucleases. The most popular pair of scissors consists of a simple piece of RNA and an unremarkable enzyme that cleaves double-stranded DNA. The positioning partner consists of clustered regularly interspaced short palindromic repeats (CRISPR), sections of nucleic acid embedded within the bacterial chromosome, archeological remnants of previous viral infections. The actual scissors are Cas proteins, enzymes known as nucleases that are present in a great abundance of cells. Naturally designed, synthesized, and created by the bacterial cell during an initial viral infection, a second round of infection by the same viral agent activates transcription of these DNA repeats into CRISPR RNA (crRNA) which seeks out and pairs with the Cas scissors to form the CRISPR/Cas gene editing tool, the genetic breakthrough technology of at least the last five decades [Citation1,Citation2]. CRISPR/Cas activity extends to gene repression, epigenetic modification, gene activation, and multiple nicking editing ().
Figure 1. The many faces of CRISPR/Cas gene editing. The activated CRISPR/Cas9 complex can modified cell behavior in a variety of ways. These include deletion of sections of genomic DNA, the insertion or replacement of segments of the genome (viewed as being traditional genomic gene editing), modification of gene expression where in the complex functions as a repressor or activator of transcription and modulation of chromatin structure using effector domains of remodeling enzymes. dCas9 refers to a Cas nine protein that is devoid of double-strand DNA breakage, while Nickase refers to a Cas9 protein where in only one of the two DNA cleavage domains of Cas9 is active. This results in the making of a single strand of the helix
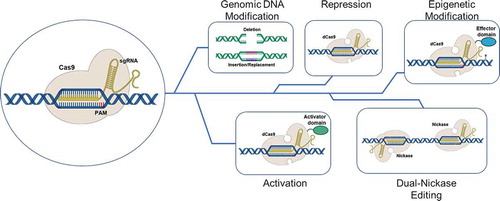
As described above, bacterial cells have evolved an adaptive immunity system which protects them against secondary infection of viruses, akin to the antibody response in humans. CRISPR/Cas is part of a prokaryotic type II immune system wherein short fragments of viral DNA are cleaved and incorporated at the CRISPR locus straddling repeat sequences that consist of DNA elements known as protospacers, short pieces of DNA derived from the corresponding parts of the invading viral genome. Upon activation, the CRISPR/Cas complex attacks the secondary invaders cleaving and disabling their DNA at predestined sites. Such cleavage and subsequent resection by cellular repair systems can block protein expression or enable the translation of truncated proteins that are not likely to be fully functional.
But the democratization or transformation of the reagents that comprise adaptive immunity in bacterial cells for use in mammalian cells is what captures the imagination of the scientific community and frankly, the public at large. Is this the genetic tool that will enable us to repair mutations accurately and restore normal function to cells that are disabled or dysfunctional? Maybe. There is no doubt that this instrument has accelerated the evolution of gene editing so quickly that we are just now beginning to understand its applications and, inevitably, its power. While CRISPR/Cas dominates the news, there are several other programmable nucleases that are used to meet the challenge of effective gene editing in eukaryotes. Zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) are functional in some applications of genetic engineering, but the versatility and ease of use is why the CRISPR/Cas system captures all the headlines [Citation3].
2. The BC (Before CRISPR) Era
As with every tool at the disposal of genetic engineers, primitive versions do exist. While no one can be sure where the original concept of changing genomic DNA with an exogenously added molecule came from, seminal experiments conducted by Sherman and his colleagues in the 1980s are certainly among the leading candidates [Citation4,Citation5]. These workers carried out their experiments in the tractable genetic system, baker’s yeast, or Saccharomyces cerevisiae because the anticipated frequency with which the genome would be changed was anticipated to be extremely low; if genome editing events were to be identified, thousands of samples needed to be screened. In almost all these studies, the tool of choice was a single-stranded DNA oligonucleotide, synthetic DNA that was introduced/transfected into the cells using liposomes or electroporation. This field was generally known as single-agent gene editing and laid somewhat dormant until work began in the late 1990s that transitioned the approach into mammalian cells [Citation6,Citation7]. Numerous groups began to unravel and elucidate the mechanism of action and regulatory circuitry of how these tools worked, and how they were controlled in mammalian cells [Citation8–12]. One of the most important observations was that double-stranded DNA breaks in the chromosome enhance the capacity of the single-stranded DNA oligonucleotide to repair point mutations and patch short deletions [Citation10,Citation13,Citation14]. This enhancement was due to the activation of the DNA damage response pathway, which responds when DNA damage occurs in the chromosomes. DNA breakage was achieved by introducing anticancer drugs or radiation into/onto cultured cells, resulting in random scission throughout the genome [Citation15].
While these experiments were critical in potentiating human gene editing, it was simply impractical to induce random double-strand breaks in patient samples even if a certain percentage of the cells enabled the designated site-specific repair of the targeted mutation. Thus, there was a need for a new category of genetic tools, molecular scissors that could be designed to cut at a specific site at or near the mutation; hence, the evolution of programmable nucleases began. It is from this basket of tools, that CRISPR-directed gene editing [Citation16–20], in combination with single-stranded DNA oligonucleotides, has emerged bringing hope for the treatment of inherited diseases and certain forms of cancer.
3. Does CRISPR/Cas have a role as a direct or indirect therapeutic modality in T cell-based therapy?
There is a plethora of outstanding and very informative reviews written by experts on the development of immunotherapies and the evolution of CAR-associated strategies; I will not attempt to contribute to that literature. Rather, I will spend the rest of the review discussing how CRISPR/Cas systems are being used in conjunction with immunotherapy, offer perhaps a new application with some very basic molecular data, and then provide some perspective and caution based on our own experience with working with this remarkable genetic tool.
Because immunotherapy can empower the immune system to find and attack cancer cells, it is clear to all of us that it is uniquely positioned to, and in fact already is, have a significant impact on cancer treatment. Perhaps the most popular form of immunotherapy is based on cellular adaptive approaches or T cell transfer, where the patient’s T cells are collected from blood and new types of membrane-bound receptors known as chimeric antigen receptors (CARs) are engineered on the cell surface. With proper planning and design, CARs enable T cells to recognize uniquely defined antigens/targets. Variations of the CAR T strategy, including T cell receptor therapy and Natural Killer (NK) cell therapy, are maneuvering through clinical trials. In some cases, the re-engineering process already employed CRISPR/Cas so that human gene editing has already had an impact on the world of immunotherapy. CRISPR/Cas systems have also been used to create models for drug discovery in cells bearing AML-FLT3 mutations [Citation21] and a CRISPR/Cas system demonstrated reversion of adverse effects of BCR/ABL in a xenograft model of CML [Citation22], to name but a few examples.
As predicted, however, there are some challenges even for CAR T cell therapy in liquid tumor indications. In some patients, CAR T cells do not expand enough, nor do they persist, especially with patients with CLL [Citation23,Citation24]. In elegant studies, Fraietta et al. [Citation25] suggested that intrinsic defects in the T cells themselves appear to prevent significant levels of therapeutic activation and expansion in vivo. A second type of challenge is fundamental: the poor success in the expansion of the patients’ T cells that have been collected from the blood and introduced into large-scale culture, in preparation for (re) infusion. An even more practical problem sometimes arises; the process by which a patient’s acquired T cells are expanded often takes so much time, the therapeutic benefit to the patient is systematically reduced; it just takes too long to get the cells back. And, despite the optimism surrounded treatment of solid tumors, there is still a paucity of data supporting the notion that CAR T cell therapy will be successful for solid tumors.
4. Innovative applications of CRISPR/Cas
The versatility of the CRISPR/Cas system enables unique and innovative applications. As mentioned above, although some patients cannot receive genetically modified CAR T cells, is it possible to create a bank of universal T cells from healthy donors that would be widely available to cancer patients? And could these cells provide independence from the graft-versus-host reaction? We do know that the off-the-shelf CAR T cell products could also induce a graft-versus-host disease response and ultimately rejection by the host. With the precision and efficiency of CRISPR/Cas activity engineered into these molecules, it might be possible that programmable nucleases could knock out genes that facilitate immune rejection. The alpha-beta T cell receptor might be a useful target for knockout as well as the elimination of beta-2 microglobulin, which serves as a subunit of HLA-I protein. Ren et al. [Citation26] published provocative preclinical studies indicating that multiple CRISPR/Cas gene editing activity, known as multiplexing, was in fact effective in knocking out endogenous TCR in CAR T cells, yet retained antitumor activity without inducing GVHD. Other targets for genetic knockout could include stimulatory NK cell ligands or the nonclassical HLA class I molecules on allogenic T cells.
For quite some time it has been clear that diminishment in T cell function seems to be controlled in part by multiple negative checkpoint regulators, such as PD-1. Other inhibitory receptors are outlined in ; TIM-3, LAG-3, TIGIT and CTLA-4 are known to fulfill a similar function after tumor antigen interaction [Citation27,Citation28]. It is possible that these receptors work independently, yet collaborate to suffocate immune response [Citation29]. An obvious solution to the problem would be to provide checkpoint inhibitors in association with the CAR T cell therapy, but systemic delivery has now been shown to induce adverse reactions [Citation24,Citation30].
Table 1. Outline of Target Sites for Cas9-mediated Immune Checkpoint Inhibition in T-cells
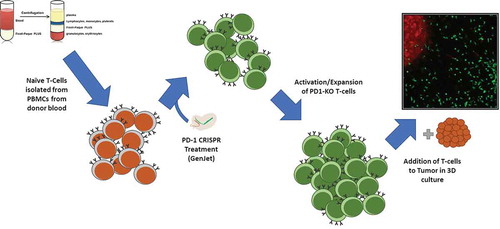
So, using our toolmaking skills, we designed, synthesized, and created a CRISPR/Cas genetic tool to evaluate the hypothesis that a knockout of PD-1 in human T cells would enhance activity. It was a simple, broad-based experiment to illustrate the application of CRISPR/Cas in current context of this review. Primary blood mononuclear cells were isolated from donor blood, and naïve T cells were further isolated from the PBMCs using FACS. It was found that gene knockout prior to T cell activation and expansion was necessary for continued expansion of the activated T cells. If PD-1 knockout was performed after the cells underwent initial expansion, overactivation occurred that led to mass T cell-mediated cytotoxicity throughout the population. The entire culture, including the feeder layer, was killed in 6–72 hours. When targeting was performed prior to T cell activation, however, the T cells were able to be expanded indefinitely using standard methods.
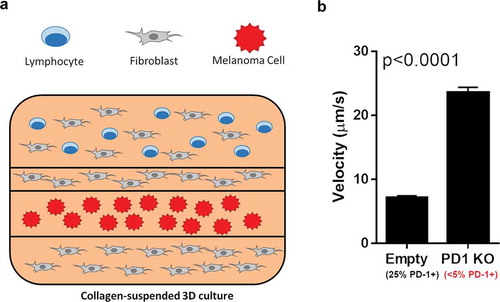
Using this second experimental approach (), primary T cells were transfected with a PD-1-targeting CRISPR/Cas9 construct. After transfection, the cells were activated with a mild activation factor (activation with CD3 also led to complete population cytotoxicity) and expanded for several days. After targeting, the cells showed a 5x decrease in surface PD-1. The effectiveness of these T-cells in tumor recognition was then measured by the metric of tumor infiltration velocity [Citation31]. Briefly, the T cell population was labeled with a tracer dye, then layered on a collagen gel containing both unlabeled fibroblasts and melanoma cells labeled with a different dye. These 3D cultured could then be live imaged over the following 72 hours, allowing for tracking of the movements of the T cells and calculation of their tumor infiltration velocity.
Figure 2. Method for knockout of PD-1 in T cells. PBMCs were isolated from patient blood samples, and T cells were further isolated from the PBMC population by FACS. Isolated naïve T cells were targeted via liposome-based transfection with a CRISPR construct plasmid and a GFP reporter. Successfully transfected cells were pooled and activated with a weak activation factor, and allowed to expand for several days. After expansion and confirmation of cytotoxic potential, the activated cells, tumor infiltration capacity was analyzed in 3D culture
shows the outcome of the experiment. The PD-1 knockout cells, once expanded and recovered from targeting, maintained their cytotoxic capacity. When the PD-1 knockout cells were exposed to the tumor cells via the 3D model, tumor infiltration speed had remarkably increased by over 3x. Time-lapse videos of these cultures are included as Supplemental Materials 1 & 2. What is also interesting, however, is the apparent reaction of the tumor cells to the increased T cell infiltration. In the PD-1 knockout experiment, tumor cell migration appears to be much higher than was seen in the control. In both videos, tumor cell migration can be seen. In the PD-1 knockout cells, this migration appears to occur significantly faster and to a higher extent. From our perspective, this provides foundational observations that will need a more scientifically dedicated series of experiments to establish a positive correlation. But it certainly is intriguing that genetic manipulation of other loci could improve the effectiveness of modified T cells for immunotherapy.
Figure 3. 3D culture-based infiltration assay outline. (A) Fluorescence-labeled T cells were suspended in a collagen matrix, layered with fibroblasts and labeled melanoma cells in the displayed layered 3D culture. This culture was live imaged over the course of 72 hours, with the migration vector and velocity of the T-cells recorded. (B) After 72 hours, the overall velocity of the T cells with and without PD-1 knockout were analyzed. Mock-transfected cells exhibited surface PD-1 on approximately 25% of cells, while PD1-KO cells showed surface PD-1 on less than 5%. An increase in tumor infiltration velocity of over 3x was seen between mock-transfected and PD-1 KO T cells
5. Expert Opinion: CRISPR in the scientific world
Thus, it is now possible, even feasible, to utilize CRISPR/Cas to disable the genes within the allogenic T cell to bypass the immunosuppressive reaction? Perhaps. In the reengineering of any mammalian cell, it is likely that only a certain percentage of the cells will be disabled, and in fact if multiple targets are designated for destruction, a mosaic population will be generated. Some cells will contain the knockout of gene A, others might harbor the knockout of gene B, perhaps there will be a certain percent with the designated is a knockout of A + B + C, etc.; there is no guarantee that multiplexing is equally effective in each targeted cell, and this has plagued a variety of cancer screens. It is also important to note that genetically modified cells tend to change the growth patterns, even if the targeted and disabled gene is not generally associated with cell proliferation. Bialk et al. [Citation32] demonstrated that the knockout of NRF2, a transcriptional regulator involved in chemoresistance, but not known to affect viability, led to a diminishment in cell cycle time, a slowing of the cell division process. Reducing the rate of replicating cells can often be disadvantageous for combinatorial standard of care such as chemotherapy. It is important to understand that the negative effects of CRISPR-directed gene editing are not only on the human genome; metabolic pathways, some unknown to be interactive, can also be affected by even a slight reduction in gene expression patterns, and lead to unexpected cell behavior. Gao et al. [Citation33] Provided an excellent overview of the challenges facing the implementation of CRISPR/Cas gene editing engineered T cell therapy. These authors emphasize that the manufacturing complexity of biomolecular tools and the infiltration of engrafted cells are simple yet foundational obstacles the clinical application. Thus, more work is required at the molecular, the cellular, and the production levels to ensure that the full potential of CRISPR/Cas activity is realized.
Challenges in the implementation of clinical applications cannot be overstated. Even with the CAR and TCR therapy approachs, two therapeutic modalities already in use, there is significant inconsistency in the mode of delivery. Delivery of biotherapeutics, specifically gene medicines, has been a significant barrier for over 25 years, and while important progress has certainly been made, there is no universal vector that can accomplish efficient and effective delivery of biological payloads. Workers have naturally chosen human viruses that have been modified not to cause their inherent infection, and as predicted, cell penetration and tropism remain high. However, these viral vectors can still induce immune response despite a quarter century of molecular biology activity to reduce this response. Finally, much publicity surrounds CRISPR activity in terms of potential capacity to modify non-targeted sites. We believe this is unavoidable since CRISPR/Cas is a natural biological activity. However, while other sites will unavoidably be targeted, the choice to use gene editing will probably involve risk analyses; whether the potential for genetic modulation at secondary sites, occurring at very low frequencies, outweighs the potential for long-term benefits. Just like pharmaceuticals, CRISPR-directed gene editing is not without its side effects.
CRISPR in the material world
As exciting as all the remarkable advancements in the science of CRISPR have been, there is a reality to the application of gene editing in patients that is often overlooked. Many scientists would probably agree that it is too soon to move CRISPR-directed gene editing into clinical application, but there are hundreds of clinical protocols in process at the FDA, and several clinical trials at mid-phase [Citation34]. At the present time, there are no reports of severe adverse effects when CRISPR is used as the main genetic tool, although more traditional gene therapy approaches continue to present problematic outcomes [Citation35]. The advancement of gene editing into the clinic is no easy matter, and one of the most important hurdles has nothing to do with science but lives in the material world. Obtaining the proper licenses to produce a therapy that can be used in a designated patient population is complicated, expensive and, at best, confusing. The well-known CRISPR patent wars between Massachusetts Institute of Technology and the University of California at Berkeley have dominated the scientific legal docket. While this has certainly been entertaining to those of us who utilize this tool daily, the complex nature of licensing for use of this technology could prohibit important studies from going forward. Frankly, right now, it is not exactly clear how many licenses one would need to execute and produce a therapeutic product that can reach the people who need the treatment the most [Citation36].
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosure
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Hsu PD, Lander ES, Zhang F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell. [Internet]. 2014;157:1262–1278. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867414006047
- Sontheimer EJ, Barrangou R. The Bacterial Origins of the CRISPR Genome-Editing Revolution. Hum Gene Ther. [Internet]. 2015;26:413–424. .
- Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. [Internet]. 2018;9:1911. Available from: http://www.nature.com/articles/s41467-018-04252-2
- Yamamoto T, Moerschell RP, Wakem LP, et al. Strand-specificity in the transformation of yeast with synthetic oligonucleotides. Genetics. [Internet]. 1992;131:811–819. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1205094%7B&%7Dtool=pmcentrez%7B&%7Drendertype=abstract
- Moerschell RP, Tsunasawa S, Sherman F. Transformation of yeast with synthetic oligonucleotides. Proc Natl Acad Sci U S A. [Internet]. 1988;85:524–528. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=279583%7B&%7Dtool=pmcentrez%7B&%7Drendertype=abstract
- Aarts M, te Riele H. Progress and prospects: oligonucleotide-directed gene modification in mouse embryonic stem cells: a route to therapeutic application. Gene Ther. [Internet]. 2011;18:213–219. Available from: http://www.nature.com/articles/gt2010161
- Parekh-Olmedo H, Ferrara L, Brachman E, et al. Gene therapy progress and prospects: targeted gene repair. Gene Ther. [Internet]. 2005;12:639–646. Available from: http://www.nature.com/articles/3302511
- Dekker M, Brouwers C, Aarts M, et al. Effective oligonucleotide-mediated gene disruption in ES cells lacking the mismatch repair protein MSH3. Gene Ther. [Internet]. 2006;13:686–694. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16437133
- Papaioannou I, Disterer P, Owen JS. Use of internally nuclease-protected single-strand DNA oligonucleotides and silencing of the mismatch repair protein, MSH2, enhances the replication of corrected cells following gene editing. J Gene Med. [Internet]. 2009;11:267–274. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19153972
- Morozov V, Wawrousek EF. Single-strand DNA-mediated targeted mutagenesis of genomic DNA in early mouse embryos is stimulated by Rad51/54 and by Ku70/86 inhibition. Gene Ther. [Internet]. 2008;15:468–472. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18079752
- Igoucheva O, Alexeev V, Scharer O, et al. Involvement of ERCC1/XPF and XPG in Oligodeoxynucleotide-directed Gene Modification. Oligonucleotides. [Internet]. 2006;16:94–104. .
- Olsen PA, Randol M, Luna L, et al. Genomic sequence correction by single-stranded DNA oligonucleotides: role of DNA synthesis and chemical modifications of the oligonucleotide ends. J Gene Med. [Internet]. 2005;7:1534–1544. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16025558
- Ferrara L, Parekh-Olmedo H, Kmiec EB. Enhanced oligonucleotide-directed gene targeting in mammalian cells following treatment with DNA damaging agents. Exp Cell Res. [Internet]. 2004;300:170–179. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15383324
- Wu X-S, Xin L, Yin W-X, et al. Increased efficiency of oligonucleotide-mediated gene repair through slowing replication fork progression. Proc Natl Acad Sci U S A. [Internet]. 2005;102:2508–2513. Available from: http://www.pnas.org/content/102/7/2508.short
- Ferrara L, Kmiec EB. Camptothecin enhances the frequency of oligonucleotide-directed gene repair in mammalian cells by inducing DNA damage and activating homologous recombination. Nucleic Acids Res. [Internet]. 2004;32:5239–5248. Available from: http://nar.oxfordjournals.org/content/32/17/5239.long
- Jinek M, Chylinski K, Fonfara I, et al. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. [Internet]. 2012;337:816–821. .
- Jinek M, East A, Cheng A, et al. RNA-programmed genome editing in human cells. Elife. [Internet]. 2013;2:e00471. Available from: http://elifesciences.org/content/2/e00471.abstract
- Cho SW, Kim S, Kim JM, et al. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. [Internet]. 2013;31:230–232. Available from: http://www.nature.com/articles/nbt.2507
- Cong L, Ran FA, Cox D, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. [Internet]. 2013;339:819–823. .
- Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826.
- Rivera-Torres N, Banas K, Kmiec EB. Modeling pediatric AML FLT3 mutations using CRISPR/Cas12a- mediated gene editing. Leuk Lymphoma. [Internet]. 2020;61:3078–3088. .
- García-Tuñón I, Hernández-Sánchez M, Ordoñez JL, et al. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget. [Internet]. 2017;8:26 027–26040. .
- Porter DL, Hwang W-T, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. [Internet]. 2015;7:303ra139–303ra139. .
- Salas-Mckee J, Kong W, Gladney WL, et al. CRISPR/Cas9-based genome editing in the era of CAR T cell immunotherapy. Hum Vaccin Immunother. [Internet]. 2019;15:1126–1132. .
- Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. [Internet]. 2018;24:563–571. Available from: http://www.nature.com/articles/s41591-018-0010-1
- Ren J, Zhang X, Liu X, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. [Internet]. 2017;8:17002–17011. .
- Turnis ME, Andrews LP, Vignali DAA. Inhibitory receptors as targets for cancer immunotherapy. Eur J Immunol. [Internet]. 2015;45:1892–1905. .
- Sakuishi K, Apetoh L, Sullivan JM, et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. [Internet]. 2010;207:2187–2194. Available from: https://rupress.org/jem/article/207/10/2187/40732/Targeting-Tim3-and-PD1-pathways-to-reverse-T-cell
- Odorizzi PM, Wherry EJ. Inhibitory Receptors on Lymphocytes: insights from Infections. J Immunol. [Internet]. 2012;188:2957–2965. .
- Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. Longo DL, editor. N Engl J Med. [Internet]. 2018;378:158–168. .
- Zhang T, Somasundaram R, Berencsi K, et al. Migration of cytotoxic T lymphocytes toward melanoma cells in three-dimensional organotypic culture is dependent on CCL2 and CCR4. Eur J Immunol. [Internet]. 2006;36:457–467. .
- Bialk P, Wang Y, Banas K, et al. Functional Gene Knockout of NRF2 Increases Chemosensitivity of Human Lung Cancer A549 Cells In Vitro and in a Xenograft Mouse Model. Mol Ther - Oncolytics. [Internet]. 2018;11:75–89. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2372770518300263
- Gao Q, Dong X, Xu Q, et al. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T‐cell therapy. Cancer Med. [Internet]. 2019;8:4254–4264. .
- NIH Clinical Trials Database [Internet]. cited 2021 Mar 3. Available from: https://clinicaltrials.gov/.
- Snyder J, Falcetti C, Goldberg I. bluebird bio Announces Temporary Suspension on Phase 1/2 and Phase 3 Studies of LentiGlobin Gene Therapy for Sickle Cell Disease (bb1111) [Internet]. 2021. Available from: https://investor.bluebirdbio.com/news-releases/news-release-details/bluebird-bio-announces-temporary-suspension-phase-12-and-phase-3. Accessed: March 1, 2021.
- Kmiec E, Marron J. Potential Inequities in New Medical Technologies. Sci Am. [Internet]. 2020 Mar 28. Available from: https://blogs.scientificamerican.com/observations/potential-inequities-in-new-medical-technologies/