
ABSTRACT
Introduction
β-thalassemia is one of the most common inherited monogenic diseases. Many patients are dependent on a lifetime of red blood cell (RBC) transfusions and iron chelation therapy. Although treatments have a significant impact on quality of life (QoL), life expectancy, and long-term health outcomes have improved in recent decades through safer RBC transfusion practices and better iron chelation strategies. Advances in the understanding of the pathology of β-thalassemia have led to the development of new treatment options that have the potential to reduce the RBC transfusion burden in patients with transfusion-dependent (TD) β-thalassemia and improve QoL.
Areas covered
This review provides an overview of currently available treatments for patients with TD β-thalassemia, highlighting QoL issues, and providing an update on current clinical experience plus important practical points for two new treatments available for TD β-thalassemia: betibeglogene autotemcel (beti-cel) gene therapy and the erythroid maturation agent luspatercept, an activin ligand trap.
Expert opinion
Approved therapies, including curative gene therapies and supportive treatments such as luspatercept, have the potential to reduce RBC transfusion burden, and improve clinical outcomes and QoL in patients with TD β-thalassemia. Cost of treatment is, however, likely to be a significant barrier for payors and patients.
1. Introduction
β-thalassemia, an autosomal recessive disorder arising from single mutations that reduce the expression of β-globin, is one of the most common monogenic inherited diseases worldwide [Citation1–3]. While β-thalassemia is prevalent in more than 60 countries across the world, most patients originate from Southeast Asia, the Middle East, and the Mediterranean [Citation2,Citation4,Citation5]. β-thalassemia is characterized by the absence (β0) or reduced synthesis (β+) of the β-globin subunit of adult hemoglobin, causing an imbalance in the ratio of α-globin and β-globin chains, reducing hemoglobin production, and promoting ineffective erythropoiesis [Citation6]. The premature destruction of red blood cell (RBC) precursors in the bone marrow and other extramedullary sites results in chronic anemia, reduced tissue oxygenation, and increased erythropoietin synthesis, which cause a wide range of clinical consequences, including bone changes, splenic enlargement, enlarged bone marrow, growth retardation, organ damage, vascular dysfunction, and jaundice [Citation7–10].
Conventional treatments for patients with transfusion-dependent (TD) β-thalassemia are regular RBC transfusions plus iron chelation therapy (ICT) to correct anemia and reduce iron accumulation (). Hematopoietic stem cell transplantation (HSCT), the only curative option, is only available for a very limited subgroup of patients with a suitable matched donor () [Citation8,Citation11,Citation12]. Patients with more clinically severe disease, known as TD β-thalassemia, often present with symptoms very early in life (before the age of 2 years), require lifelong disease management, and are dependent on regular RBC transfusions in order to prevent organ damage or early death [Citation8,Citation9]. Widespread adoption of safe RBC transfusion practices and improved ICT options have led to a longer life expectancy and improvements in the quality of life (QoL) for these patients; however, outcomes are still suboptimal for many [Citation13,Citation14]. TD β-thalassemia impairs the QoL of patients significantly compared with the general population [Citation15,Citation16]. Children and young adults are particularly affected as they lose important time in school with their peers due to regular hospital visits for treatments and appointments with numerous specialists [Citation17]. Their frequent and invasive supportive treatments also cause long-term psychosocial effects.
Figure 1. Currently approved treatments for transfusion-dependent β-thalassemia
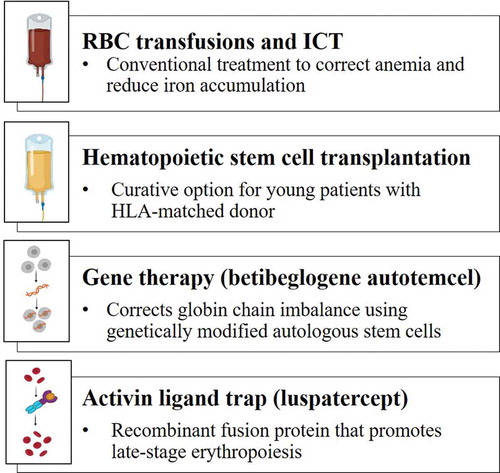
Advances in the understanding of the pathogenesis of β-thalassemia have led to the development of several new treatment approaches and novel therapies that address the underlying cause of the disease. These can broadly be divided into three groups: those that aim to correct the imbalance in globin chain synthesis (i.e. gene therapy or gene editing), those that improve late-stage erythropoiesis (e.g. activin ligand traps, JAK2 inhibitors), and those that reduce iron accumulation (e.g. hepcidin-like molecules, transmembrane protease serine 6 inhibitors) [Citation11,Citation18]. This review will focus on the potential to improve QoL for patients with TD β-thalassemia by summarizing current management recommendations and focusing on clinical use of two novel treatments that have recently been approved (gene therapy and the activin ligand trap, luspatercept; ). These treatments have the potential to dramatically improve QoL outcomes for patients, necessitating guidance for physicians as we await their inclusion in treatment guidelines [Citation8,Citation19–21].
2. Current treatment strategies and their effect on QoL
2.1. Conventional treatment approach
The management of patients with TD β-thalassemia is complex. Regular monitoring and intervention are required to manage complications, including cardiovascular disease, liver disease, bone disease, endocrine disease, and growth abnormalities [Citation8]. Infection risk is increased due to both the disease and treatment. Patients often require specialist care for psychological support and issues related to fertility and pregnancy.
Treatment options for TD β-thalassemia generally include supportive care (e.g. RBC transfusion, ICT) and potentially curative therapies (e.g. HSCT, gene therapy). Splenectomy can reduce transfusion requirements and the risk of iron overload [Citation22]. Due to the risk of complications, splenectomy use has decreased in recent decades and is not generally recommended for patients with TD disease, provided that patients receive adequate RBC transfusions and ICT, as discussed below [Citation8].
2.2. RBC transfusions and ICT
Patients who have TD β-thalassemia require lifelong treatment with regular RBC transfusions every 2–5 weeks to maintain pretransfusion hemoglobin levels of 9–10.5 g/dL [Citation8]. The current international recommendations for clinical practice to ensure safe blood transfusions for thalassemia patients are summarized in . Patients who are regularly transfused face a number of important and potentially serious complications, including increased risk of viral infection from contaminated blood [Citation23,Citation24] and alloimmunization [Citation8,Citation25]. It is therefore important to screen all donated blood for viral contamination and use Rh- and K-matched donor blood for all transfusions. In countries where TD β-thalassemia is endemic, the need for regular, safe blood supplies is a major healthcare burden [Citation14,Citation26–28]. Even in relatively wealthy nations, the demand for blood supplies is a significant burden for healthcare systems. In Greece, for example, it was reported that the 4506 patients with hemoglobinopathies in the Greek national registry between 1997 and 2010 utilized 18% of the country’s total supply of RBCs [Citation28]. Uncertainties in securing adequate blood supply on an individual basis cause significant psychological burden on patients. Therefore, treatment strategies that reduce RBC transfusion requirements would have benefits for national healthcare services as well as for individual patients. Moreover, the SARS-CoV-2 pandemic has led to global shortages in blood donation and supplies since the beginning of 2020 [Citation29–33], leaving patients with β-thalassemia particularly vulnerable.
Table 1. Current international recommendations for safe blood transfusion of patients with transfusion-dependent β-thalassemia
Despite improvements in screening for pathogens and efforts to minimize allogenic reactions, both remain as consequences of transfusion and have potential to increase treatment burden [Citation14,Citation34]. These treatment burdens, while not universal, clearly affect health-related quality of life (HRQoL) in RBC transfusion recipients. Numerous longitudinal studies from the international Thalassemia Clinical Research Network have demonstrated impaired HRQoL in patients with β-thalassemia compared with the general US population [Citation34]. The burden of RBC transfusion and related morbidity can be safely assumed to contribute to these impairments. HRQoL worsened with increasing transfusion frequency and in line with the average distance traveled each month to receive blood, with school functioning being the domain most impacted [Citation35].
Patients receiving regular RBC transfusions must adhere to an adequate ICT regimen to prevent complications or death due to iron toxicity. Iron can accumulate at a rate of 0.3–0.6 mg/kg per day in transfused patients and cannot be excreted by the body [Citation8]. The accumulated iron has a number of toxic effects that include damage to the heart and liver, growth retardation, endocrine dysfunction, and osteoporosis, all of which may contribute to decreased QoL [Citation36].
While improved chelation regimens have reduced mortality due to cardiac failure, the leading cause of premature death in patients with TD β-thalassemia, patients continue to experience complications and stress from cardiac-related mortality [Citation37]. In addition, long-term health issues that negatively affect patients’ QoL, such as bone pain, osteoporosis, liver disease, liver cancer, and anxiety, have emerged [Citation15,Citation38–41]. While children receiving optimal transfusion and chelation have better QoL, over time this worsens due to the numerous complications related to iron overload and repeated transfusions [Citation36].
ICT is very effective in preventing iron overload if patients comply with and adhere to treatment, but these regimens can be very demanding of patient time, patient health, and healthcare resources and result in reduced QoL [Citation2,Citation9,Citation14,Citation42]. Three iron chelator drugs are currently available: deferoxamine, deferiprone, and deferasirox. Deferoxamine is administered by subcutaneous or intramuscular injection or intravenous infusion on a 5–7 days per week dosing schedule [Citation43]. The two oral agents, deferiprone and deferasirox, are taken three times daily [Citation44,Citation45]. Suggested guidelines for ICT regimens are summarized in . Non-oral ICT can substantially affect HRQoL due to frequent, often daily, administration through a subcutaneous pump for at least 8–10 consecutive hours. This generally occurs during the night, with the potential to impair sleep, and is also associated with pain at the site of injection [Citation46,Citation47]. Oral ICT is preferred by patients, which leads to better adherence [Citation48–50] and (possibly) QoL [Citation14]. In a recent meta-analysis (2008–2016) that included 2961 patients with TD β-thalassemia, those receiving deferoxamine and combination therapy had poorer HRQoL status across all eight domains of the 36-item Short Form (SF-36) health survey than those receiving deferasirox [Citation48]. In addition, a new film-coated formulation of deferasirox that can be taken with a meal, rather than on an empty stomach, has advantages in adherence, satisfaction/preference, and patient concerns compared to the dispersible formulation [Citation51]. The dose of ICT must be adjusted for each patient and careful regular monitoring is required to manage the adverse effects, such as nausea, joint pain, kidney injury, and agranulocytosis, which may be associated with treatment and may compromise adherence [Citation8,Citation9,Citation17,Citation52,Citation53]. Additionally, over time, comorbidity due to iron overload and transfusion complications is likely to lead to polypharmacy, which has been shown to be a major issue for patients who do not adhere to ICT [Citation47]. Other concerns with ICT use reported by both adhering and non-adhering patients include the high cost and the lack of immediate results from ICT [Citation47].
Table 2. Standard ICT regimens for transfusion-dependent β-thalassemia patients receiving regular blood transfusions
3. HSCT
Allogeneic transplantation with HLA-matched hematopoietic stem cells is the only curative treatment option for patients with TD β-thalassemia. This approach, however, is only suitable for young patients who have an HLA-matched donor, usually a sibling [Citation54]. Consensus statements currently recommend offering HSCT to patients younger than14 years of age who have a suitable HLA-matched donor [Citation55,Citation56]. It is estimated that > 70% of patients do not have a suitable sibling HLA-matched donor, although non-sibling-related donors may also be available, particularly in populations where large families and consanguineous marriages are common [Citation54,Citation57,Citation58]. Some evidence suggests that good outcomes can be achieved with unrelated donors, provided that donor and recipient are closely HLA-matched; in some cases, outcomes with unrelated donors were comparable to those achieved using matched sibling donors [Citation8,Citation57,Citation59–62]. Other alternative HSCT strategies, such as HLA-antigen-mismatch donors [Citation58], pretransplant immunosuppression [Citation61], or double-unit unrelated cord blood transplantation [Citation62], may further broaden the safety and/or applicability of HSCT. In addition, advances in preimplantation genetic diagnosis have enabled the selection of an HLA-matched embryo to produce a sibling donor, although the ethics of producing ‘savior siblings’ have been questioned [Citation63,Citation64].
Patients who undergo conventional HSCT before 14 years of age have a very low (< 10%) procedure-related mortality rate and the disease-free survival rate is at least 80% [Citation55,Citation56]. In a 30-year follow-up study (median follow up 11 years), Caocci and colleagues reported equivalent long-term survival in 258 HSCT-treated (including 161 [63.4%] patients < 16 years; 30-year overall survival rate 82.6 ± 2.7%) and 258 conventionally treated patients (85.3 ± 2.7%); patients with unrelated donors experienced shorter survival compared with those who had a sibling donor [Citation65]. Follow-up after HSCT should include careful monitoring for engraftment and possible relapse, infection, and graft-versus-host disease (GVHD), particularly in the first year after transplantation [Citation8]. Thereafter, patients should be monitored for potential long-term complications, including iron overload, endocrine impairment, and issues related to growth and development [Citation8]. Post-HSCT management of iron overload may require ICT or phlebotomy but should not be initiated until the graft is stable and immunosuppressive therapy has been discontinued. Issues related to endocrine dysfunction and infertility should be managed by a specialist.
After 20 years, improved HRQoL has been reported in patients who had HSCT compared with those who had conventional treatment [Citation35,Citation66]. The socioeconomic benefits to patients include less time off for medical appointments, improved education and job prospects, and better daily QoL. Studies have shown that HSCT improves HRQoL and is particularly valuable for young children as it offers the potential for lifelong freedom from RBC transfusions and ICT, allowing for increased continuity in education and social situations [Citation66,Citation67].
Even though the costs of HSCT and follow-up vary between countries [Citation68], a successful transplant can be considered cost-effective compared with the costs of a lifetime of conventional treatment [Citation9,Citation56,Citation69]. Disadvantages of HSCT include the intensive myeloablative and immunosuppressive conditioning that are required before the procedure, the risk of acute or chronic GVHD, negative effects on fertility, and the limited number of patients for whom the treatment is an option due to the lack of a suitable donor [Citation8,Citation9,Citation39]. In a recent systematic review, studies reported both acute and chronic GVHD having a negative effect on HRQoL scores [Citation67]. Treatment of GVHD may also worsen QoL due to side effects including seizures and posterior leukoencephalopathy, although the replacement of cyclosporine by mycophenolate mofetil has led to a reduction in these complications [Citation70,Citation71].
School function was the most severely affected domain in an Indian study of HSCT versus conventional therapy [Citation35], and there was no improvement in social domain scores in many of the studies in the systematic review by Badawy and colleagues [Citation67]. Both issues potentially stem from the need for prolonged hospitalization and isolation to avoid infection [Citation67]. In addition, impaired growth and increased likelihood of being overweight may add to existing stigma related to the disease [Citation67,Citation72].
4. Beti-cel gene therapy
Gene therapy is a novel and potentially curative treatment strategy for TD β-thalassemia that has been designed to correct the underlying globin chain imbalance, thus improving the production of functional hemoglobin, erythropoiesis, and chronic anemia. Significantly, gene therapy does not require a matched donor. The aim of gene therapy is to enable patients to achieve long-term transfusion independence [Citation5,Citation39,Citation73]. Patients undergo autologous hematopoietic stem cell mobilization and harvesting, followed by intensive myeloablative conditioning and immunosuppression. The autologous stem cells obtained are then transduced outside the body with self-inactivating lentiviral vectors that insert a gene construct containing the globin gene and other genetic elements required for gene expression. The transduced stem cells are then reinfused into the patient where the modified cells replicate, repopulate the blood compartment, and facilitate normal hemoglobin synthesis ().
Figure 2. Gene therapy clinical process
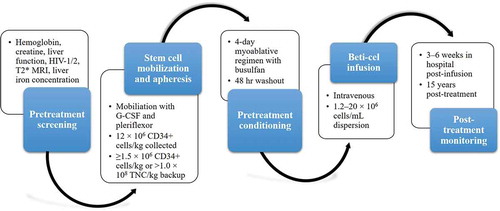
The first gene therapy, betibeglogene autotemcel (beti-cel; bluebird bio), is currently in clinical trials in the USA for patients with TD β-thalassemia aged ≤ 50 years for whom HSCT is appropriate but no HLA-matched donor is available. Beti-cel is a genetically modified autologous CD34+ cell enriched population that contains hematopoietic stem cells transduced with a lentiviral vector encoding the βA-T87Q-globin gene [Citation75]. Patients undergo mobilization and apheresis to collect CD34+ stem cells and a full myeloablative conditioning regimen with busulfan prior to beti-cel infusion.
Lentiviral vector-based gene therapy has been successful in several animal models of β-thalassemia and proof-of-principle studies in an adult patient with TD β-thalassemia [Citation76–80]. In two non-randomized open-label phase 1/2 studies, mobilized autologous CD34+ cells were obtained from 22 patients with TD β-thalassemia and transduced with a lentiviral vector encoding adult hemoglobin A with a T87Q substitution. The transduced stem cells were then re-infused after the patients had undergone myeloablative conditioning with busulfan [Citation81]. Both studies (NCT01745120, NCT02151526) reported increased levels of hemoglobin A and a sustained reduction in transfusion requirement. The adverse events reported were similar to those associated with autologous stem cell transplantation and no clonal dominance related to lentiviral vector integration was observed [Citation81]. At a median of 26 months after receiving gene therapy, all except 1 of 13 patients with non-β0β0 genotypes had achieved transfusion independence and biological markers indicated that ineffective erythropoiesis had been corrected [Citation81]. Although the benefits of treatment were most pronounced in patients with less severe genotypes, 9 patients with β0β0 genotypes achieved a 73% reduction in median annual transfusion volume and 3 of these patients discontinued RBC transfusions [Citation81].
The clinical efficacy and safety of beti-cel in patients with non-β0β0 and β0β0 genotypes have been demonstrated in the HGB-212 and HGB-207 phase 3 studies (NCT02906202, NCT03207009) [Citation82]. After a median 24.3-month follow-up, 30 of 34 (88.2%) patients treated in phase 3 were transfusion independent [Citation83]. Of 7 evaluable β0β0 patients, 6 (85.7%) achieved transfusion independence (defined as Hb ≥ 9 g/dL without packed RBC transfusions for ≥ 12 months) for > 20.6 months [Citation83]. The safety profile of beti-cel treatment was similar to that seen following myeloablative conditioning with busulfan [Citation83] . Following initial positive results, the phase 3 studies were expanded to include pediatric patients; interim results in the 28 pediatric and adolescents enrolled in these studies up to March 2021 show that pediatric patients achieve transfusion independence at comparable rates (91%) to adults with a similar safety profile [Citation84,Citation84].
In an ongoing 13-year follow-up study (LT-303) involving 32 patients from the phase 1/2 and phase 3 studies, transfusion independence was achieved by 14 of 22 patients from the phase 1/2 studies (64%; 12 in the parent study, 2 in LT-303) and 9 of 10 (90%) treated in the phase 3 Northstar-3 study [Citation81,Citation84]. In addition, patients in the phase 3 study continue to experience improved erythropoiesis, reduced iron burden, and cessation of ICT (median follow-up 14.4 months) [Citation85]. Thus, while most patients treated with beti-cel achieved long-term transfusion independence, it is important to note that some patients may still require intermittent transfusions, albeit at a lower rate than before treatment.
An analysis of data from 110 patients followed up for up to 5 years in 6 studies of beti-cel in TD β-thalassemia, sickle cell disease, and cerebral adrenoleukodystrophy found that gene therapy carried no risk of graft rejection or GVHD, and no clinically relevant clonal dominance or lentiviral vector-mediated replication competent lentivirus were observed [Citation86]. A temporary hold of clinical trials of beti-cel in β-thalassemia, due to investigations into the development of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) in 2 patients with sickle cell disease in a related clinical trial, has been lifted; the MDS diagnosis has been revised to TD anemia and the AML case was reported to be unlikely related to treatment [Citation87]. While provisional approval by the European Medicines Agency (EMA) was given in 2019, bluebird bio has recently announced they will no longer be focusing on gaining further market access for beti-cel in Europe due to payor challenges [Citation88]. Another gene therapy involving intrabone administration of hematopoietic stem cells transduced with the lentiviral vector GLOBE is also undergoing clinical development and has shown promising early results in a phase 1/2 clinical trial (NCT02453477) [Citation89].
4.1. Beti-cel gene therapy: benefits and challenges
Gene therapy has important potential to expand the number of patients who can receive potentially curative treatment for TD β-thalassemia as it avoids the need for an HLA-matched donor, there is no risk of GVHD, and long-term immunosuppression is not required. The potential risks associated with lentiviral gene therapy include viral toxicity and the activation of oncogenes causing tumor development and transmission in the germ line. None of these issues have been observed in clinical trials of beti-cel in patients with TD β-thalassemia, but long-term data are needed and will be provided by the long-term follow-up study LT-303 [Citation82,Citation83,Citation86]. Additionally, the toxicity of the myeloablative conditioning regimen and difficulties in adequate hematopoietic stem cell mobilization and harvesting can be compromised by the suppressive effect of long-term transfusions and ICT on the bone marrow [Citation18].
The impact of gene therapy on patient QoL remains to be fully determined and should be further evaluated in future clinical trials of beti-cel. However, data from the phase 3 Hgb-207 (Northstar-2) and Hgb-212 (Northstar-3) studies showed that patients under 18 years of age who achieved transfusion independence had an improved EuroQoL 5 dimensions (Youth) (EQ-5D-Y) score (67 [range 50–96] versus 92.5 [Citation85–95]) after 12 months [Citation67,Citation90]. In addition, HSCT has been shown to have a positive impact on both physical and emotional aspects of HRQoL in patients with TD β-thalassemia and, given GVHD was the most common cause of impaired HRQoL in these patients, a similar or greater benefit would be expected from gene therapy [Citation67].
Gene therapies have an extremely high initial cost, which will be challenging for health authorities and payors and may not be affordable in developing countries [Citation91]. The hematopoietic stem cell mobilization/apheresis protocol and the myeloablative conditioning regimen are very resource intensive and the requirement of individualized treatment prevents any economies of scale in production. As has been previously described for HSCT, gene therapy could theoretically be cost-effective if it allows patients to achieve transfusion independence and avoid the high monthly costs associated with long-term transfusions, ICT, and related healthcare expenses; however, the cost of gene therapy is considerably higher than that of HSCT. A recent health economic analysis of beti-cel therapy versus standard of care for TD β-thalassemia in France showed that beti-cel was cost-effective with an incremental cost-effectiveness ratio of EUR 49 per quality-adjusted life-year (QALY), rising to EUR 427/QALY when indirect costs such as unemployment and loss of productivity were included [Citation92].
4.2. Beti-cel gene therapy: clinical practice points
Beti-cel gene therapy can only be administered in an experienced treatment center by a multidisciplinary team experienced in delivering HSCT and treating patients with TD β-thalassemia. Beti-cel is prepared on an individual patient basis; it is for autologous use only and should be administered once as an intravenous infusion following full myeloablative conditioning.
Eligible patients ≥ 12 years of age must have hemoglobin levels ≥ 11 g/dL for 30 days prior to mobilization and during myeloablation; creatinine clearance ≤ 70 mL/min/1.73 m2; normal liver function tests; negative serology HIV-1/2; and cardiac T2*-weighted magnetic resonance imaging (T2*MRI) ≥ 10 msec. If MRI liver iron concentration is ≥ 15 mg/g, liver biopsy should be performed to confirm absence of fibrosis, cirrhosis, or active hepatitis. Negative serum pregnancy tests must be provided before mobilization and conditioning, and before beti-cel infusion. Beti-cel is not recommended for women who are breastfeeding. Ova and semen cryopreservation are recommended before treatment. Anti-retroviral medications and/or hydroxyurea should be stopped ≥ 1 month prior to and until ≥ 7 days after conditioning and ICT must be stopped ≥ 7 days before conditioning. It is recommended that patients receive prophylaxis to prevent veno-occlusive disease and prophylaxis against seizures should also be considered.
Stem cell mobilization is performed approximately 2 months prior to beti-cel infusion using granulocyte-macrophage-colony stimulating factor and pleriflexor followed by apheresis to collect a minimum of 12 × 106 CD34+ cells/kg. Repeat cycles of mobilization and apheresis may be required to collect sufficient cells. A back-up collection of ≥ 1.5 × 106 CD34+ cells/kg or > 1.0 × 108 TNC/kg (bone marrow harvest) is required. Pretreatment conditioning should only begin once the complete beti-cel doses have been received on site. Six days before beti-cel infusion, a full 4-day pretreatment myoablative conditioning regimen with busulfan is required, followed by a 48-hour ‘washout’ period. Beti-cel is administered intravenously in a hospital setting. Prior to initiating infusion, reconfirm that the identity of the patient matches the name on the infusion bag(s). Beti-cel (1.2–20 × 106 cells/mL dispersion) should be infused within 4 hours of thawing and each infusion bag must be infused within 30 minutes. Patients should remain in the hospital for 3–6 weeks for monitoring.
Standard procedures for patient monitoring and management following HSCT must be followed after administration of beti-cel, including for thrombocytopenia and bleeding. Patients must be tested annually for leukemia or lymphoma for 15 years post treatment. The long-term effects of gene therapy on iron already accumulated in the liver and heart are not clear and patients may therefore need to continue or restart ICT following the procedure. Guidelines are needed for ICT post-gene therapy; current recommendations based on consensus opinion are shown in [Citation93].
Table 3. Recommendations for post-gene therapy ICT
It is important to provide adequate patient/parent/carrier counseling and education regarding the risks and complexity of this treatment prior to initiating gene therapy. It is particularly important that patients and their families understand the short-term risks associated with the intensive conditioning regimen that is required and the possible need to continue ICT after gene therapy. Patient enrollment in the product registry for 15 years following treatment should also be discussed.
5. Luspatercept
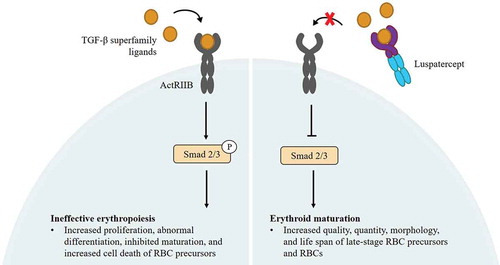
Luspatercept (Bristol Myers Squibb) is a novel recombinant fusion protein that binds transforming growth factor beta (TGF-β) superfamily ligands to inhibit aberrant Smad2/Smad3 signaling and promote late-stage erythropoiesis [Citation94,Citation95] (). Luspatercept was approved for the treatment of anemia in adult patients with TD β-thalassemia by the US FDA in November 2019 and by the EMA in June 2020 [Citation96,Citation97]. In healthy volunteers, luspatercept treatment increased hemoglobin levels and was safe and well tolerated at effective dose levels [Citation98]. A multicenter, open-label, randomized dose-finding phase 2 study (NCT01749540, NCT02268409) demonstrated that luspatercept at 0.2–1.25 mg/kg subcutaneously (s.c.) every 3 weeks for at least 5 cycles, was effective and well tolerated in 64 adults with TD or non-TD β-thalassemia [Citation98]. A reduction in RBC transfusion burden of ≥ 20% over a 12-week period was achieved by 26 of 32 TD patients (81%) in the study; the most common Grade 1 or 2 adverse events included bone pain, headache, and myalgia [Citation99].
Figure 3. Schematic representation of the mechanism of action of luspatercept
These findings led to the initiation of the pivotal randomized, double-blind, placebo-controlled, multicenter BELIEVE phase 3 study (NCT02604433) that confirmed the efficacy and safety of luspatercept at 1.0–1.25 mg/kg s.c. every 21 days for at least 48 weeks, in adults with TD β-thalassemia [Citation100]. The proportion of patients in the BELIEVE study who achieved ≥ 33% in RBC transfusion burden during Weeks 13–24, with a reduction of ≥ 2 RBC units (primary endpoint) was significantly greater with luspatercept compared with placebo (21.5% versus 4.5%, respectively; P < 0.001). During any 12-week period, a significantly greater proportion of patients receiving luspatercept achieved ≥ 33% or ≥ 50% reduction in RBC transfusion burden compared with placebo [Citation100]. Subgroup analysis of the BELIEVE study has shown that clinically meaningful reductions in transfusion burden were seen across all genotypes, though response rates were lower in patients with the most severe (β0β0) disease [Citation101]. Luspatercept was well tolerated; adverse events that were more common with luspatercept than placebo were transient bone pain, arthralgia, dizziness, hypertension, and hyperuricemia [Citation100]. Thrombotic adverse events were reported in 8 patients (4%) treated with luspatercept, mostly in splenectomized patients with known thrombotic risk factors.
Recent follow-up analyses from the BELIEVE study have confirmed that the benefit of luspatercept on transfusion burden is sustained over time. Patients who continued luspatercept treatment experienced sustained reductions in RBC units transfused and transfusion visits for at least 2 years in a longitudinal analysis of the open-label phase of the study [Citation102,Citation103]. As of 1 July 2019, the median duration of treatment for patients in the luspatercept and placebo groups was 119.1 weeks and 74.7 weeks, respectively. In patients treated with luspatercept, reductions in transfusion burden (≥ 2 RBC units) from baseline of ≥ 33% and ≥ 50% during any 48-week period were achieved by 27.7% and 12.5% of patients, respectively, compared with 1.8% and 0.9% of patients in the placebo group, respectively [Citation103]. In patients initially randomized to the placebo arm who crossed over to luspatercept treatment during the open-label phase, 18.5% achieved ≥ 33% reduction in RBC and 9.8% achieved reductions of transfusion burden of ≥ 50% during Weeks 13–24 after crossover [Citation103]. In addition, compared with placebo, luspatercept treatment was associated with a significant reduction in serum iron levels, liver iron concentration, and myocardial iron levels during the first 96 weeks of treatment [Citation104]. Longer term use of luspatercept was associated with an increasing number of patients with serum ferritin levels < 1000 µg/L and trends toward a reduction in overall use of ICT and decreasing daily doses of deferasirox [Citation105].
5.1. Luspatercept: benefits and challenges
Compared with gene therapy and HSCT, drugs that correct ineffective erythropoiesis, such as luspatercept, are less expensive and may therefore be more accessible to patients. Health economic studies will be needed to demonstrate the long-term cost-effectiveness of this new treatment. The long-term impact of luspatercept on transfusion burden (e.g. volume of RBC units transfused, frequency of transfusion visits) and comorbidities in the real-world setting is not yet known. Observational studies of clinical practice patterns should provide valuable insights on these important issues. Preliminary evidence from the BELIEVE study indicates that luspatercept provides a sustained reduction in serum ferritin levels, however, changes to cardiac and hepatic iron overload take time. Thus, patients will require close follow-up (e.g. T2* MRI). The anticipated benefits of luspatercept in reducing transfusion burden would be expected to translate into positive benefits on patients’ physical and mental well-being. Preliminary data from the BELIEVE trial suggest luspatercept maintains HRQoL and improves physical functioning (as measured by the generic SF-36 health survey Physical Functioning and Physical Component Summary [PCS] questionnaire) compared with placebo [Citation106]. Patients who achieved clinical benefit with luspatercept treatment were also more likely to experience meaningful improvement in Physical Functioning and PCS compared with placebo. Long-term QoL outcomes studies are needed to confirm this effect. In addition, the long-acting nature of luspatercept in conjunction with the associated reductions in transfusion burden and ICT use could potentially ameliorate some of the QoL burden of conventional therapy over time, by improving adherence and reducing transfusion-related complications.
5.2. Luspatercept: clinical practice points
Luspatercept should be administered s.c. once every 21 days [Citation107]. The recommended starting dose is 1.0 mg/kg (maximum injected volume per site 1.2 mL) injected into the upper arm, thigh, or abdomen. A treatment response to luspatercept is defined as a reduction in RBC transfusion burden of at least one third (33%) after two consecutive doses. If no treatment response is seen at the starting dose, the dose should be increased to 1.25 mg/kg (maximum dose). If a patient loses their treatment response after initially responding to the starting dose, the dose of luspatercept can be increased to 1.25 mg/kg.
Prior to each dose of luspatercept, it is essential to assess the patient’s hemoglobin level. If a transfusion is needed before starting treatment, the pre-transfusion hemoglobin level should be used to assess the starting dose of luspatercept [Citation106]. During treatment, hemoglobin levels should be assessed regularly. If a patient’s hemoglobin level increases > 2 g/dL within 3 weeks of a dose of luspatercept in the absence of a transfusion, the next dose of luspatercept should be reduced by 1 step (e.g. a 1.0 mg/kg to 0.8 mg/kg or 1.25 mg/kg to 1.0 mg/kg). If a patient’s hemoglobin levels increase to > 11.5 g/dL with no transfusions in the previous 3 weeks, the next luspatercept dose should be delayed until their hemoglobin level is < 11.0 g/dL and a single-step dose reduction in luspatercept dose should be considered as described above. If a patient experiences a loss of response to luspatercept, the dose can be increased to 1.25 mg/dL after excluding potential causes such as a bleeding episode. If no treatment response is seen after 3 doses (e.g. 9 weeks) at the maximum dose level (1.25 mg/kg), treatment should be discontinued. No dose adjustments are required for patients with mild to moderate hepatic or renal impairment.
Adverse events, such as transient bone pain, arthralgia, dizziness, hypertension, and hyperuricemia, are usually transient and should be managed with prophylaxis. Patients, especially those with risk factors and previous splenectomy, should be actively monitored for thrombosis [Citation108]. If patients develop persistent, high-grade (Grade 3 or above) adverse events related to treatment, the next dose of luspatercept should be delayed until the toxicity has resolved or significantly improved; treatment should be restarted at the previous dose (or a lower dose should be considered). Treatment should be discontinued if a patient experiences a Grade 3–4 hypersensitivity reaction.
6. Conclusions and challenges
HSCT continues to play a valuable role in the treatment of TD β-thalassemia, and improved techniques, such as the use of matched unrelated donors, are improving the safety and applicability of this approach. Emerging clinical evidence suggests that the newly available treatment options, gene therapy and luspatercept, are likely to further improve long-term clinical outcomes, reduce transfusion burden, improve long-term health, maintain HRQoL, and increase life expectancy for patients with TD β-thalassemia. Significant challenges remain with newly approved treatments, including barriers to treatment for patients and payors (e.g. the cost and cost-effectiveness of treatment, availability, and accessibility of treatment), the risks associated with gene therapy (i.e. intensive conditioning regimen, stem cell procurement), the impact of new treatments on future need for ICT and long-term QoL, and provision of appropriate patient education and counseling.
7. Expert opinion
Undoubtedly the long-term survival and QoL have significantly improved over the past decades for patients with TD β-thalassemia. The increased availability of safe, screened Rh- and K-matched blood, and oral ICT drugs have transformed and lengthened lives for many patients. However, the majority of patients with β-thalassemia live in resource-constrained areas of the world; for these patients, access to adequate transfusions and appropriate ICT as well as the life-long burden of this disease remains challenging. Even in patients achieving effective control with these therapies, comorbidities increase over time and lead to reductions in life expectancy and QoL. As a potentially curative treatment, HSCT plays an important role for selected patients. New techniques improving the safety and efficacy of matched family and unrelated donors will continue to expand the candidates for HSCT. Despite these advances, however, not all patients will be eligible for HSCT. For these reasons, new treatment approaches that can reduce the long-term need for RBC transfusions and/or the dose/frequency of ICT are being developed.
Among the emerging new treatment options, gene therapy has the potential to increase the number of patients who can receive curative treatment that can offer lifelong independence from RBC transfusions without the need for a matched sibling donor. For some countries, such as those with limited availability of donated blood, the wider availability of a treatment that reduces the need for RBC transfusions will be important. However, the fact that gene therapy is a very expensive and specialized treatment limits its use in many areas of the world. Health economic studies are needed to demonstrate the cost/benefit of gene therapy over a patient’s life. In addition, the long-term safety of gene therapy will need to be carefully monitored for the potential, though currently unsubstantiated, activation of oncogenes.
The approval of luspatercept has led to the availability of a new treatment for patients with TD β-thalassemia and may provide long-term sustained reductions in transfusion requirements and the need for ICT. These benefits should translate into improved long-term outcomes for patients and associated improvements in QoL. Long-term data from studies such as the phase 3 BELIEVE trial, which has a 5-year follow-up, are now emerging and will provide valuable insights in this respect, but initial signs are that luspatercept can provide sustained reductions in both transfusion burden and the frequency of transfusions.
The cost of treatment directly impacts access to new therapies and is a key consideration in the management of patients with TD β-thalassemia that will greatly impact the use of these treatments in the management of this disease. It is clear that there will be a need for updated treatment recommendations from national thalassemia societies/healthcare agencies to guide the integration of the novel therapies into standard care pathways and to inform physicians, payors, and patients on what to expect from, and how to prepare for, gene therapy and luspatercept treatment in clinical practice. Accordingly, the Thalassemia International Federation has recently updated its guidance on the use of gene therapy and luspatercept [Citation8]. Several other novel treatments for TD β-thalassemia in clinical development aim to improve iron metabolism and reduce iron accumulation and erythropoiesis (e.g. gene editing, hepcidin-like molecules, TMPRSS6 inhibitors, ferroportin inhibitors), extend RBC survival (e.g. allosteric activators of pyruvate kinase, inhibitors of phosphodiesterase-9), or develop alternative sources of RBCs that may overcome infection risk or be compatible for patients with alloimmunization (e.g. in vitro RBC cultures, erythropoietic stem cells derivatives, induced pluripotent stem cells, genetically modified somatic cells). Several of these novel agents are expected to be approved within the next 5 years and collectively these new treatments will improve long-term outcomes for patients with β-thalassemia.
List of Abbreviations
AML: acute myeloid leukemia
CPD-A: citrate-phosphate-dextrose-adenine
EMA: European Medicines Agency
G-CSF: granulocyte colony-stimulating factor
GVHD: graft-versus-host disease
HIV: human immunodeficiency virus
HRQoL: health-related quality of life
HSCT: hematopoietic stem cell transplant
ICT: iron chelation therapy
MDS: myelodysplastic syndromes
MRI: magnetic resonance imaging
PCS: Physical Component Summary
QALY: quality-adjusted life-year
QoL: quality of life
RBC: red blood cell
s.c.: subcutaneously
SF-36: 36-item Short Form
TD: transfusion-dependent
TGF-β: transforming growth factor beta
TNC: total nucleated cell count
Article highlights
The life expectancy and quality of life of patients with transfusion-dependent (TD) β-thalassemia have been improved by advances in the safety of red blood cell (RBC) transfusions and better iron chelation strategies, but lifelong transfusions and chelation therapy are still a huge social and economic burden for patients
The first treatments addressing the underlying pathology of TD β-thalassemia have recently been approved: the gene therapy betibeglogene autotemcel (beti-cel) and luspatercept, an activin ligand trap that reduces ineffective erythropoiesis
Gene therapy using globin lentiviral vectors has the potential to provide curative treatment for patients with TD β-thalassemia, offering the possibility of long-term transfusion independence previously only available following hematopoietic stem cell transplantation
Luspatercept improves late-stage erythroid maturation and has been shown to significantly reduce RBC transfusion requirements in patients with TD β-thalassemia with a notable proportion of patients in clinical trials achieving RBC transfusion independence
Practical guidance on the use of gene therapy and luspatercept in real-world clinical practice are provided as consensus expert recommendations on the use of these new treatments become available
Declaration of interest
AT Taher reports consultancy fees from Agios, Celgene (BMS), Ionis Pharmaceuticals, Novartis , and Vifor Pharma; and grants for research from Celgene (BMS), Ionis Pharmaceuticals, Novartis , and Vifor Pharma. A Kattamis reports advisory board, steering committee, research funding, speaker’s fees, and travel support from BMS; speaker’s fees, advisory board, steering committee, and research funding from Novartis; speaker’s fee, advisory board, and steering committee for Apopharma, Chiesi, and CRISPR Therapeutics/ Vertex; and advisory board and steering committee for Agios, Ionis, and Vifor Pharma. MD Cappellini reports advisory board for BMS, Novartis, Novo Nordisk, Sanofi Genzyme, and Vifor Pharma. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The authors received editorial and writing support from Jacqueline Moy, PhD, from Excerpta Medica, funded by Bristol Myers Squibb, Princeton, New Jersey. The authors are fully responsible for all content and editorial decisions for this article.
Additional information
Funding
References
- Rund R, Rachmilewitz E. Medical progress. β-thalassemia. N Engl J Med. 2005;15353(11):1135–1146. PubMed PMID: 16162884.
- Modell B, Darlisson M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Heal Organ. 2008;86(6):480–487. PubMed PMID: 18568278.
- HbVar. A database of human hemoglobin variants and thalassemias. [cited 2021 May 20]. Available from: http://globin.cse.psu.edu/hbvar/menu.html.
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331–4336. PubMed PMID: 20233970.
- Amjad F, Fatima T, Fayyaz T, et al. Novel genetic therapeutic approaches for modulating the severity of thalassemia. Biomed Rep. 2020;13(5):48. PubMed PMID: 32953110.
- Weatherall DJ, Clegg JB. The thalassaemia syndromes. 4th ed. Oxford: Wiley-Blackwell; 2013.
- Nienhuis AW, Nathan DG. Pathophysiology and clinical manifestations of the β thalassemias. Cold Spring Harb Perspect Med. 2012;2(12):a011726. PubMed PMID: 23209183.
- Cappellini MD, Farmakis D, Porter J, et al. 2021 Guidelines for the management of transfusion dependent thalassaemia (TDT). 4th ed. Nicosia, Cyprus: Thalassaemia International Federation; 2021.
- Capellini MD, Porter JB, Viprakasit V, et al. A paradigm shift on β-thalassemia treatment: how will we manage this old disease with new therapies?. Blood Rev. 2018;32(4):300–311. PubMed PMID: 29455932.
- Casu C, Pettinato M, Liu A, et al. Correcting β-thalassemia by combined therapies that restrict iron and modulate erythropoietin activity. Blood. 2020;136(17):1968–1979. PubMed PMID: 32556142.
- Bou-Fakhedrin R, Tabbikha R, Daadaa H, et al. Emerging therapies in β-thalassemia: toward a new era in management. Expert Opin Emerg Drugs. 2020;25(2):113–122. PubMed PMID: 32249632.
- Baronciani D, Pilo F, Lyon-Caen S, et al. Hematopoietic stem cell transplantation in thalassemia major report from the EBMT hemoglobinopathy registry. Blood. 2011;118(21):905.
- Cunningham MJ. Update on thalassemia: clinical care and complications. Pediatr Clin North Am. 2008;55(2):447–460. PubMed PMID: 18381095.
- Shah FT, Sayani F, Trompeter S, et al. Challenges of blood transfusions in β-thalassemia. Blood Rev. 2019;37:100588. PubMed PMID: 31324412.
- Gan GG, Hue YL, Sathar J, et al. Factors affecting quality of life in adult patients with thalassaemia major and intermedia. Ann Acad Med Singap. 2016;45(11):520-523. PubMed PMID: 27922147.
- Cappellini MD, Kattamis A, Viprakasit V, et al. Quality of life in patients with β-thalassemia: a prospective study of TD and non-transfusion dependent patients in Greece, Italy, Lebanon, and Thailand. Am J Hematol. 2019;94(10):E261–264. PubMed PMID: 31321793.
- Dhirar N, Khandekar J, Bachani D, et al. Thalassemia major: how do we improve quality of life?. Springerplus. 2016;5(1):1895. PubMed PMID: 27843752.
- Motta I, Bou-Fakhedrin R, Taher AT, et al. Beta thalassemia: new therapeutic options beyond transfusion and iron chelation. Drugs. 2020;80(11):1053–1063. PubMed PMID: 32557398.
- United Kingdom Thalassaemia Society. 2016. Standards for the clinical care of children and adults with thalassemia in the UK. 3rd ed. [cited 2021 May 20]. Available from: www.ukts.org/standards/Standards-2016final.pdf.
- Children’s Hospital & Research Center Oakland. 2012. [cited 2021 May 20]. Available from: www.thalassemia.com/documents/SOCGuidelines2012.pdf.
- Sayani F, Warner M, Wu J, et al. G2009. [cited 2021 May 20]. Available from: www.thalassemia.ca/wp-content/uploads/Thalassemia-Guidelines_LR.pdf.
- Piga A, Serra M, Longo F, et al. Changing patterns of splenectomy in transfusion-dependent thalassemia patients. Am J Hematol. 2011;86(9):808–810. PubMed PMID 21850661.
- Mancuso A, Sciarrino E, Renda MC, et al. A prospective study of hepatocellular carcinoma incidence in thalassaemia. Haemoglobin. 2006;30(1):119–124. PubMed PMID: 16540424.
- Mahmoud RA, El‑Mazary AA, Khodeary A, et al. Seroprevalence of hepatitis C, hepatitis B, cytomegalovirus, and human immunodeficiency viruses in multitransfused thalassemic children in upper Egypt. Adv Hematol. 2016;2016:9032627. PubMed PMID: 26989417.
- Tormey CA, Hendrickson JE. Transfusion-related red cell alloantibodies: induction and consequences. Blood. 2019;133(17):1821–1830. PubMed PMID: 30808636.
- Weatherall DJ. The evolving spectrum of the epidemiology of thalassemia. Hematol Oncol Clin North Am. 2018;32(2):165–175. PubMed PMID: 29458724.
- Basak Aliz S, Yildirim M, Atunay H, et al. Effects of the problems faced by patients with thalassemia during supply of blood and blood transfusion. Vox Sang. 2012;103(S1):77.
- Politis C. Haemoglobinopathies: genetic and clinical aspects with an impact on blood transfusion. Vox Sang. 2013;105(S1):58–59.
- Shander A, Goobie SM, Warner MA, et al. Essential role of patient blood management in a pandemic: a call for action. Anesth Analg. 2020;131(1):74–85. PubMed PMID: 32243296.
- Loua A, Kasilo OMJ, Nikiema JB, et al. Impact of the COVID‐19 pandemic on blood supply and demand in the WHO African Region. Vox Sang. 2021. PubMed PMID: 33529421. DOI:https://doi.org/10.1111/vox.13071.
- Raturi M, Kusum A. The blood supply management amid the COVID-19 outbreak. Transfus Clin Biol. 2020;27(3):147–151. PubMed PMID: 32386966.
- Mohammadi S, Tabatabaei Yazdi SM, Eshghi P, et al. Coronavirus disease 2019 (COVID-19) and decrease in blood donation: experience of Iranian blood transfusion organization (IBTO). Vox Sang. 2020;115(7):595–596. PubMed PMID: 32270880.
- Franchini M, Farrugia A, Velati C, et al. The impact of the SARS-CoV-2 outbreak on the safety and availability of blood transfusions in Italy. Vox Sang. 2020;115(8):603–605. PubMed PMID: 32240543.
- Betts M, Flight PA, Paramore LC, et al. Systematic literature review of the burden of disease and treatment for transfusion-dependent beta-thalassemia. Clin Ther. 2020;42(2):322–337. e2. PubMed PMID: 31882227.
- Chordiya K, Katewa V, Sharma P, et al. Quality of life (QoL) and the factors affecting it in transfusion-dependent thalassemic children. Indian J Pediatr. 2018;85(11):978–983. PubMed PMID: 29752583.
- Patel S, Swaminathan VV, Mythili VS, et al. Quality matters - hematopoietic stem cell transplantation versus transfusion and chelation in thalassemia major. Indian Pediatr. 2018;55(12):1056–1058. PMID: 30745477.
- Borgna-Pignatti C, Rugolotto S, Stefano D, et al. Survival and complications in patients with thalassaemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187–1193. PubMed PMID: 15477202.
- Pinto VM, Poggi M, Russo R, et al. Management of the aging beta-thalassemia transfusion-dependent population – the Italian experience. Blood Rev. 2019;38:100594. PubMed PMID: 31416718.
- Porter J. Beyond transfusion therapy: new therapies in thalassemia including drugs, alternate donor transplant, and gene therapy. Hematology Am Soc Hematol Educ Program. 2018;2018(1):361–370. PubMed PMID: 30504333.
- Haidar R, Musallam KM, Taher AT, et al. Bone disease and skeletal complications in patients with β thalassemia major. Bone. 2011;48(3):425–432. PubMed PMID: 21035575.
- Haines D, Martin M, Carson S, et al. Pain in thalassaemia: the effects of age on pain frequency and severity. Br J Haematol. 2013;160(5):680–687. PubMed PMID: 23278768.
- Amid A, Saliba AN, Taher AT, et al.Thalassaemia in children: from quality of care to quality of
- Novartis Pharmaceuticals Corporation. Desferal®deferoxamine mesylate prescribing information. [cited 2021 Mar 17]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/016267s044lbl.pdf. http://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=016267
- ApoPharma Inc. Ferriprox® (deferiprone) tablets for oral use prescribing information. [cited 2021 Mar 17]. Available from: http://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021825
- Novartis Pharmaceuticals Corporation. Exjade® (deferasirox) tablets, for oral suspension prescribing information. [cited 2021 Mar 17]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021882s024lbl.pdf
- Goulas V, Kouraklis-Symeonidis A, Manousou K, et al. A multicenter cross-sectional study of the quality of life and iron chelation treatment satisfaction of patients with transfusion-dependent beta-thalassemia, in routine care settings in Western Greece. Qual Life Res. 2021;30(2):467–477. PMID: 32920766.
- Sidhu S, Kakkar S, Dewan P, et al. Adherence to iron chelation therapy and its determinants. Int J Hematol Oncol Stem Cell Res. 2021;15(1):27–34. PMID: 33613898.
- Arian M, Mirmohammadkhani M, Ghorbani R, et al. Health-related quality of life (HRQoL) in beta-thalassemia major (beta-TM) patients assessed by 36-item short form health survey (SF-36): a meta-analysis. Qual Life Res. 2019;28(2):321–334. PMID: 30194626.
- Cappellini MD, Bejaoui M, Agaoglu L, et al. Prospective evaluation of patient-reported outcomes during treatment with deferasirox or deferoxamine for iron overload in patients with β-thalassemia. Clin Ther. 2007;29(5):909–917. PubMed PMID: 17697909.
- Olivieri N, Brittenham G. Final results of the randomised trial of deferiprone and deferoxamine. Blood. 1997;90:264a.
- Taher AT, Origa R, Perrotta S, et al. Patient-reported outcomes from a randomized phase II study of the deferasirox film-coated tablet in patients with transfusion-dependent anemias. Health Qual Life Outcomes. 2018;16(1):216. PMID: 30453981.
- Maggio A, Kattamis A, Felisi M, et al. Evaluation of the efficacy and safety of deferiprone compared with deferasirox in paediatric patients with transfusion-dependent haemoglobinopathies (DEEP-2): a multicentre, randomised, open-label, non-inferiority, phase 3 trial. Lancet Haematol. 2020;7(6):e469–e478. PubMed PMID 32470438.
- Nafea OE, Zakaria M, Hassan T, et al. Subclinical nephrotoxicity in patients with beta-thalassemia: role of urinary kidney injury molecule. Drug Chem Toxicol. 2019;5:1–10. PubMed PMID 31905029.
- Sadelain M, Boulad F, Galanello R, et al. Therapeutic options for patients with severe β-thalassaemia: the need for globin gene therapy. Hum Gene Ther. 2007;18(1):1–9. PubMed PMID 17173507.
- Angelucci E, Matthes-Martin S, Baronciani D, et al. EBMT inborn error and EBMT paediatric working parties. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99(5):811–820. PubMed PMID 2470059.
- Baronciani D, Angelucci E, Potschger U, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European society for blood and bone marrow transplantation hemoglobinopathy registry, 2000–2010. Bone Marrow Transplant. 2016;51(4):536–541. PumMed PMID 26752139.
- Aydogdu S, Toret E, Aksoy BA, et al. Comparison of hematopoietic stem cell transplantation results in patients with β-thalassemia major from three different graft types. Hemoglobin. 2021;45(1):25–29. PubMed PMID 33478286.
- Gaziev J, Marziali M, Isgrò A, et al. Bone marrow transplantation for thalassemia from alternative related donors: improved outcomes with a new approach. Blood. 2013;122(15):2751–2756. PubMed PMID 23963044.
- La Nasa G, Argiolu F, Giardini C, et al. Unrelated bone marrow transplantation for beta-thalassemia patients: the experience of the Italian bone marrow transplant group. Ann N Y Acad Sci. 2005;1054:186–195. PubMed PMID 16339665.
- Hongeng S, Pakakasama S, Chuansumrit A, et al. Outcomes of transplantation with related- and unrelated-donor stem cells in children with severe thalassemia. Biol Blood Marrow Transplant. 2006;12(6):683–687. PubMed PMID 16737942.
- Kharya G, Bakane AN, Rauthan AM, et al. Pretransplant myeloid and immune suppression, reduced toxicity conditioning with posttransplant cyclophosphamide: initial outcomes of novel approach for matched unrelated donor hematopoietic stem cell transplant for hemoglobinopathies. Pediatr Blood Cancer. 2021;68(4):e28909. PubMed PMID 33470527.
- Feng J, Lee V, Leung AWK, et al. Double-unit unrelated cord blood transplantation for thalassemia major: comparison with HLA-identical sibling bone marrow transplantation. Pediatr Transplant. 2021;25(3):e13901. PubMed PMID 33136320.
- Pennings G, Schots R, Liebaers I, et al. Ethical considerations on preimplantation genetic diagnosis for HLA typing to match a future child as a donor of haematopoietic stem cells to a sibling. Hum Reprod. 2002;17(3):534–538. PubMed PMID 11870098.
- Burgio GR, Nespoli L, Maccario R, et al. Conceiving a hematopoietic stem cell donor: twenty-five years after our decision to save a child. Haematologica. 2012;97(4):479–481. PubMed PMID 22492290.
- Caocci G, Orofino MG, Vacca A, et al. Long-term survival of beta thalassemia major patients treated with hematopoietic stem cell transplantation compared with survival with conventional treatment. Am J Hematol. 2017;92(12):1303–1310.
- La Nasa G, Caocci G, Efficace F, et al. Long-term health-related quality of life evaluated more than 20 years after hematopoietic stem cell transplantation for thalassemia. Blood. 2013;122(13):2262–2270. PMID: 23958950.
- Badawy SM, Beg U, Liem RI, et al. A systematic review of quality of life in sickle cell disease and thalassemia after stem cell transplant or gene therapy. Blood Adv. 2021;5(2):570–583. PubMed PMID 33496753.
- Matthes-Martin S, Pötschger U, Barr R, et al. Costs and cost-effectiveness of allogeneic stem cell transplantation in children are predictable. Biol Blood Marrow Transplant. 2012;18(10):1533–1539. PMID: 22484665.
- Weidlich D, Kefalas P, Guest JF, et al. Healthcare costs and outcomes of managing β-thalassemia major over 50 years in the United Kingdom. Transfusion. 2016;56(5):1038–1045. PMID: 27041389.
- Bernaudin F, Socie G, Kuentz M, et al. SFGM-TC. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749–2756. PubMed PMID 17606762.
- Bernaudin F, Pondarré C, Galambrun C, et al. Allogeneic/matched related transplantation for β-thalassemia and sickle cell anemia. Adv Exp Med Biol. 2017;1013:89–122. PubMed PMID 29127678.
- Rahal I, Galambrun C, Bertrand Y, et al. Late effects after hematopoietic stem cell transplantation for β-thalassemia major: the French national experience. Haematologica. 2018;103(7):1143–1149. PMID: 29599204.
- Karponi G, Zogas N. Gene therapy for β-thalassemia: updated perspectives. Appl Clin Genet. 2019;12:167–180. PubMed PMID 31576160.
- Kunz JB, Kulozik AE. Gene therapy of the hemoglobinopathies. HemaSphere. 2020;4(5):e479. PubMed PMID 32984772.
- Zynteglo PI. [cited 2021 Mar 18]. Available from: https://www.zynteglo.eu/pdf/euapi.pdf.
- Breda L, Casu C, Gardenghi S, et al. Therapeutic hemoglobin levels after gene transfer in beta-thalassemia mice and in hematopoietic cells of beta-thalassemia and sickle cells disease patients. PLoS One. 2012;7(3):e32345. PubMed PMID 22479321.
- May C, Rivella S, Callegari J, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406(6791):82–86. PubMed PMID 10894546.
- May C, Rivella S, Chadburn A, et al. Successful treatment of murine beta-thalassemia intermedia by transfer of the human beta-globin gene. Blood. 2002;99(6):1902–1908. PubMed PMID 11877258.
- Rivella S, May C, Chadburn A, et al. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101(8):2932–2939. PubMed PMID 12480689.
- Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467(7313):318–322. PubMed PMID 20844535.
- Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with TD β-thalassemia. N Engl J Med. 2018;378(16):1479–1493. PubMed PMID 29669226.
- Schneiderman J, Thompson AA, Walters MC, et al. Interim results from the phase 3 Hgb-207 (Northstar-2) and Hgb-212 (Northstar-3) studies of betibeglogene autotemcel gene therapy (lentiglobin) for the treatment of transfusion-dependent β-thalassemia. Biol Blood Marrow Transplant. 2020;26(3):S87–S88.
- Locatelli F, Kwiatkowski JL, Walters MC, et al. Betibeglogene autotemcel in patients with transfusion-dependent β-thalassemia: updated results from HGB-207 (Northstar-2) and HGB-212 (Northstar-3). [abstract]. EHA Library 2021. Abstract S266
- Kulozik AE, Thuret I, Kwiatkowski JL, et al. Interim results of betibeglogene autotemcel gene therapy in pediatric patients with transfusion-dependent β-thalassemia (TDT) treated in the phase 3 Northstar-2 and Northstar-3 studies. [abstract]. EHA Library 2021. Abstract EP1301.
- Kwiatkowski JL, Walters MC, Hongeng S, et al. Long-term efficacy and safety of betibeglogene autotemecel gene therapy for the treatment of TD β-thalassemia: results in patients with up to 6 years of follow-up [abstract]. Blood. 2020;136(suppl 1):153. ASH 2020. Abstract 153.
- Walters MC, Locatelli F, Thrasher AJ, et al. Safety of autologous hematopoietic stem cell transplantation with gene addition therapy for transfusion-dependent β-thalassemia, sickle cell disease, and cerebral adrenoleukodystrophy. Biol Blood Marrow Transplant. 2020;26(3):S38–S39.
- bluebird bio announces the lifting of FDA clinical hold for sickle cell disease and β-thalassemia studies. bluebird bio press release, 2021 Jun 7. [cited 2021 Jul 2]. Available from: https://www.businesswire.com/news/home/20210607005267/en/bluebird-bio-Announces-the-Lifting-of-FDA-Clinical-Hold-for-Sickle-Cell-Disease-and-%CE%B2-Thalassemia-Studies
- bluebird bio reports second quarter financial results and provides operational update. bluebird bio press release, 2021 Aug 9. [cited 2021 Aug 16]. Available from: https://www.businesswire.com/news/home/20210809005334/en/bluebird-bio-Reports-Second-Quarter-Financial-Results-and-Provides-Operational-Update
- Markel S, Scaramuzza S, Cicalese MP, et al. Intrabone hematopoietic stem cell therapy for adult and pediatric patients affected by transfusion-dependent β-thalassemia. Nat Med. 2019;25(2):234–241. PubMed PMID 30664781.
- Thompson AA, Walters MC, Mapara MY, et al. Resolution of serious vaso-occlusive pain crises and reduction in patient-reported pain intensity: results from the ongoing phase 1/2 HGB-206 group C study of LentiGlobin for sickle cell disease (bb1111) gene therapy [abstract]. Blood. 2020;136(suppl 1):16–17.
- Hanna E, Remuzat C, Auquier P, et al. Gene therapies development: slow progress and promising prospect. J Mark Access Health Policy. 2017;5(1):1265293. PubMed PMID 28265348.
- Undreiner L, Roze S, Caillon M, et al. Betiglogene autotemcel gene therapy (beti-cel) is cost-effective versus standard of care in patients with transfusion-dependent β thalassemia (TDT) in France [abstract]. Value Health. 2020;23(suppl 2):S413.
- Soni S. Gene therapies for TD β-thalassemia: current status and critical criteria for success. Am J Hematol. 2020;95(9):1099–1112. PubMed PMID 32562290.
- Suragani RN, Cawley SM, Li R, et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine β-thalassemia. Blood. 2014;123(25):3864–3872. PubMed PMID 24795345.
- Cappellini MD, Taher AT. The use of luspatercept for thalassemia in adults. Blood Adv. 2021;5(1):326–333. PubMed PMID 33570654.
- FDA. REBLOZYL label. Silver Spring, MD: FDA, 2019. [cited 2021 Feb 15]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761136lbl.pdf.
- European Medicines Agency (EMA). Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 28–30 April 2020. Amsterdam: EMA, 2020. [cited 2021 Feb 15]. Available from: https://www.ema.europa.eu/en/news/meeting-highlights-committee-medicinal-products-human-use-chmp-28-30-april-2020.
- Attie KM, Allison MJ, McClure T, et al. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am J Hematol. 2014;89(7):766–770. Pubmed PMID 24715706.
- Piga A, Perrotta S, Gamberini MR, et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with β-thalassemia. Blood. 2019;133(12):1279–1289. PubMed PMID 306177198.
- Cappellini MD, Viprakasit V, Taher AT, et al. A phase 3 trial of luspatercept in patients with TD β-thalassemia. N Engl J Med. 2020;382(13):1219–1231. PubMed PMID 32212518.
- Cappellini MD, Hermine O, Piga A, et al. Assessment of response to luspatercept by β-globin genotype in adult patients with β-thalassemia in the BELIEVE trial [abstract]. EHA Library. 2020. Abstract S295.
- Taher AT, Viprakasit V, Hermine O, et al. Sustained reductions in red blood cell transfusion burden and events in β-thalassemia with luspatercept: longitudinal results of the BELIEVE trial [abstract]. Blood. 2020;136(suppl 1):45–46.
- Taher AT, Viprakasit V, Cappellini MD, et al. Assessment of longer-term efficacy and safety in the phase 3 BELIEVE trial of luspatercept to treat anemia in patients with β-thalassemia [abstract]. EHA Library. 2020. Abstract EP1548.
- Porter J, Cappellini MD, Coates T, et al. Effects of luspatercept on iron overload and impact on responders to luspatercept: results from the BELIEVE trial [abstract]. Blood. 2019;134(suppl 1):2245.
- Hermine O, Cappellini MD, Taher AT, et al. Longitudinal effect of luspatercept on iron overload and iron chelation therapy (ICT) in adult patients with β-thalassemia in the BELIEVE trial [abstract]. Blood. 2020;136(suppl 1):47–48.
- Cappellini MD, Taher AT, Piga A, et al. Health-related quality of life outcomes for patients with transfusion-dependent beta-thalassemia treated with luspatercept in the BELIEVE trial. Blood. 2020;136(suppl 1):8–9.
- Reblozyl prescribing information/product label. [cited 2021 Mar 18]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761136orig2lbl.pdf.
- Cappellini MD, Taher AT. The use of luspatercept for thalassemia in adults. Blood Adv. 2021;5(1):326–333. PubMed PMID 33570654.