
ABSTRACT
Introduction
Waldenström’s macroglobulinemia (WM) is defined as a lymphoplasmacytic lymphoma (LPL) with immunoglobulin M (IgM) monoclonal gammopathy and morphologic evidence of bone marrow infiltration by LPL. Immunophenotyping and genotyping provide a firm pathological basis for diagnosis and are particularly valuable in differential diagnosis between WM and related diseases. Emerging technologies in mutational analysis present new opportunities, but challenges remain around standardization of methodologies and reporting of mutational data across centers.
Areas covered
The review provides an overview of the diagnosis of WM, with a particular focus on the role of immunophenotyping and genotyping.
Expert opinion
Demonstration of LPL with a bone marrow biopsy is essential to reach a definitive diagnosis of WM. However, MYD88L265P and a typical WM immunophenotypic profile are valuable for the differential diagnosis of WM and related diseases, such as marginal zone lymphoma, multiple myeloma, and chronic lymphocytic leukemia. These methodologies must be utilized across centers and with appropriate standards followed in the evaluation and reporting of sensitivities and specificities. The diagnostic and/or prognostic value of mutations in genes such as CXCR4 and TP53 that are currently not routinely evaluated in the diagnosis of WM should be explored.
1. Introduction
Waldenström’s macroglobulinemia (WM) is a lymphoplasmacytic lymphoma (LPL) with immunoglobulin M (IgM) monoclonal gammopathy and morphologic evidence of marrow infiltration by LPL [Citation1–3]. WM is the most common of two LPL subtypes recognized by the World Health Organization (WHO), accounting for approximately 95% of cases [Citation3]. The other subtype, non-WM LPL, includes cases with immunoglobulin G (IgG) or immunoglobulin A (IgA) monoclonal protein, non-secretory LPL, and IgM LPL without bone marrow involvement [Citation3]. Non-WM LPL cases are rare and poorly defined. WM comprises approximately 2% of all cases of non-Hodgkin’s lymphoma and has an incidence of ~4 cases per 1 million person-years [Citation4–6]. The disease is more common in males than in females, and in Caucasians compared with other races [Citation5]. Most patients with WM are aged ≥65 years and many have non-lymphoma-related comorbidities typical of the elderly population [Citation6,Citation7].
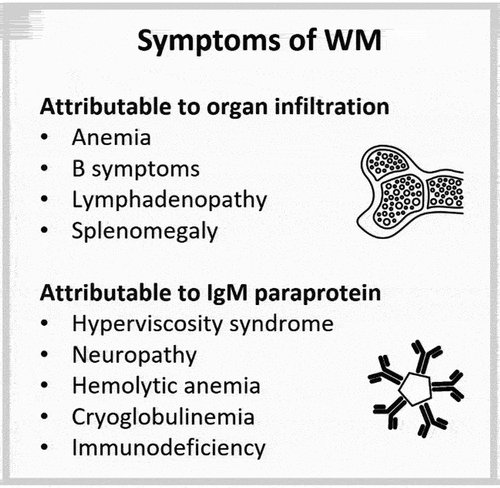
WM is associated with a heterogenous presentation, with symptoms attributable to overall disease burden as well as symptoms attributable to the concentration and/or immunological/physicochemical properties of plasma cell–secreted IgM (). Unlike other lymphomas, the malignant B-cell clone in WM undergoes plasmacytic differentiation and the extent of differentiation varies between patients. The level of IgM paraprotein correlates with the number of plasma cells rather than with the overall extent of bone marrow infiltration [Citation8,Citation9]. Hyperviscosity syndrome related to high levels of IgM is a hallmark symptom of WM, but IgM levels do not have a linear association with serum viscosity [Citation10]. Similarly, patients with low levels of IgM may experience symptoms that depend on the immunological properties of secreted IgM, such as neuropathy, hemolytic anemia, and cryoglobulinemia.
Figure 1. Symptoms of WM can be attributed to the overall disease burden and/or the IgM paraprotein. IgM: immunoglobulin M, WM: Waldenström’s macroglobulinemia.
Outcomes for patients with WM have improved over time with the introduction of new drugs and treatment regimens [Citation11–14]. Accurate and timely diagnosis is key to ensuring patients gain access to the most appropriate treatment at the right time. This review summarizes the current approach to WM diagnosis, with a particular focus on molecular diagnosis. Methods for the detection of the highly prevalent MYD88L265P mutation are considered, as well as approaches to testing for mutations in other relevant genes, such as CXCR4.
2. Clinical presentation of Waldenström’s macroglobulinemia
WM is an indolent lymphoma and about a quarter of patients with WM are asymptomatic at the time of diagnosis [Citation15]. Anemia is the primary reason for patients with WM to present to primary care or to be referred to internal medicine, being present in ~40% of symptomatic patients () [Citation15,Citation16]. B symptoms, especially weight loss, are also relatively common, reported in about a quarter of symptomatic WM patients at presentation [Citation15,Citation16]. A similar proportion of patients may present with symptoms related to hyperviscosity (arterial hypertension, bleeding, headache, cardiac insufficiency, or reduced level of consciousness) or neuropathy [Citation15,Citation16].
Figure 2. The main indication for initiation of therapy in 595 patients with WM from the Greek Myeloma Study Group database (adapted from Dimopoulos et al.) [Citation16].
![Figure 2. The main indication for initiation of therapy in 595 patients with WM from the Greek Myeloma Study Group database (adapted from Dimopoulos et al.) [Citation16].](/cms/asset/7ad17220-c413-455a-92e0-216cd66d3f28/ierr_a_2270779_f0002_oc.jpg)
3. Overview of Waldenström’s macroglobulinemia diagnosis
Differentiation of WM from other B-cell disorders () is usually achievable through correlation of clinical features, laboratory tests, bone marrow morphology, B-cell immunophenotype, and genomic profile (). Hemograms often reveal anemia in patients with WM, and more rarely leukopenia/neutropenia and/or thrombocytopenia; abnormal hemograms are often the first indication of disease in patients with asymptomatic WM. Biochemistry is usually normal, but a complete metabolic profile may reveal abnormal kidney function. While the latter is rare, with a cumulative incidence of approximately 5% it can reflect disease-specific etiologies such as amyloidosis, cast nephropathy, light chain deposition, cryoglobulinemia, and direct tumor infiltration [Citation17,Citation18]. Increased bilirubin and lactate dehydrogenase may also be found, which can be an indication of hemolysis [Citation16].
Table 1. WM differential diagnosis (adapted from Dimopoulos et al.) [Citation16].
Table 2. Essential evaluation of patients with WM (adapted from Castillo et al.) [Citation21].
Demonstration of monoclonal IgM by serum electrophoresis with immunofixation is essential for the diagnosis of WM, and evaluation of serum IgM levels is critical for prognosis and monitoring of treatment responses [Citation19,Citation20]. Funduscopic examination is recommended for patients with a serum IgM >30 g/L and for all patients with symptoms of hyperviscosity syndrome, such as oronasal bleeding and visual disturbances. A 24-hour urine analysis with urine electrophoresis and immunofixation may also be considered at diagnosis. Bence-Jones proteinuria (free monoclonal light chains in the urine) is rarer in WM than in multiple myeloma but may still be present in about a third of patients [Citation21,Citation22].
Serum free light chain (sFLC) levels and the κ/λ ratio of sFLCs may be useful indications of disease burden in WM [Citation23,Citation24]. In a study by Leleu et al., sFLC levels showed earlier response and progression than IgM or M-spike measurements in 48 patients with WM. Median κ/λ ratio was 13.5, but the 95% confidence interval (0.01–665) crossed the normal reference range (0.26–1.65) [Citation23]. There are insufficient data to recommend a role for free light chain assessment in routine response evaluation, but it is essential for patients with suspected renal complications and amyloidosis [Citation18,Citation25].
It is also important to determine serum β2 microglobulin levels because elevated levels predict poorer patient outcomes. A serum β2 microglobulin level of more than 3 mg/L is a component of the International Prognostic Scoring System for WM (IPSSWM), along with age (>65 years), hemoglobin level (≤11.5 g/dL), platelet count (≤100 × 109/L), and serum monoclonal protein concentration (>70 g/L) [Citation19].
Imaging studies are not recommended in the evaluation of asymptomatic patients, although computed tomography of the chest, abdomen, and pelvis with intravenous administration of contrast is recommended to document organomegaly and/or lymphadenopathy in patients who are considered for treatment. If organomegaly and/or lymphadenopathy are detected prior to treatment initiation, imaging during or after completion of therapy is also advised [Citation18,Citation21]. There are limited data on the utility of fluorodeoxyglucose positron emission tomography (FDG-PET) imaging in WM, although it may be applicable in some patients (for example, those with suspected histologic transformation) [Citation26,Citation27].
3.1. Histology
A diagnosis of WM requires the demonstration of morphologic marrow infiltration by LPL and this is best achieved with assessment of an adequate trephine biopsy [Citation2,Citation3,Citation18]. A trephine biopsy also provides for a better assessment of the extent of marrow disease. Arbitrary values for levels of marrow disease are no longer considered appropriate, as some symptomatic patients will have disease burdens of less than 10% [Citation25]. The value of bone marrow assessment for asymptomatic patients with IgM monoclonal gammopathy of undetermined significance (MGUS) is not well established, but patients with suspected symptomatic WM should always undergo bone marrow biopsy, including immunophenotyping and genetic assessments, independently of the level of the IgM protein [Citation18,Citation21]. IgM MGUS, which is defined as IgM monoclonal gammopathy with no evidence of bone marrow infiltration, rarely progresses to WM when no risk factors are present [Citation28–31]. Bone marrow biopsy and closer monitoring may be warranted in patients with IgM MGUS with IgM levels ≥15 g/L and/or altered free light chain assessments, because they have a greater risk of progression [Citation30,Citation31].
LPL in the bone marrow can show an interstitial, nodular, or diffuse pattern [Citation32]. Although a purely paratrabecular pattern is unusual, a combined paratrabecular and interstitial infiltration pattern is common [Citation32]. LPL cells can show differentiation from small lymphocytes with clumped chromatin, inconspicuous nucleoli, and sparse cytoplasm, to mature plasma cells, which are usually present in small numbers and may contain Dutcher bodies [Citation16,Citation21].
Plasma cell differentiation may be evident morphologically but can be further highlighted using plasma cell–specific markers, such as CD138, IRF4, and VS38c. Plasmacytoid lymphocytes are frequently detected and typically have intermediate cytologic features between small lymphocytes and plasma cells. Reactive mast cells, which may be present in the marrow microenvironment, are a characteristic feature of WM [Citation21,Citation33].
The presence of plasma cell differentiation, monotypic plasma cells, immunoglobulin inclusions, and reactive mast cells appear to be the most important morphologic features in discriminating between WM and marginal zone lymphoma (MZL) [Citation34–36].
4. Immunophenotype and other applications of flow cytometric analysis
Immunophenotyping is a powerful and potentially underutilized tool in the diagnosis of WM. In the past decade, a distinct B-cell immunophenotype for WM was identified using flow cytometric analysis [Citation37]. The WM clone expresses pan-B-cell markers (CD19, CD20, CD22, Sig) but can be distinguished from normal B cells by weak CD22 expression and homogeneous positivity for CD25 [Citation37]. Fewer than 20% of WM cases are positive for CD5, CD10, CD11c, or CD23 [Citation37,Citation38].
The B-cell compartment changes with progression from IgM MGUS through asymptomatic WM to symptomatic WM, with an increasing number of total B cells and light chain isotype positive B cells. Complete light chain restriction of the B-cell compartment appears to predict for progression in asymptomatic WM, but also overall survival in symptomatic patients [Citation37].
The plasma cell compartment is less easily distinguished from normal bone marrow plasma cells. Plasma cell clones have, by definition, restricted immunoglobulin heavy and light chain isotypes but otherwise normal antigenic expression, including positivity for CD19 and CD45, which contrasts with myeloma clones [Citation37]. In common with the B-cell compartment, the plasma cell clone changes with progression from IgM MGUS to WM [Citation37].
Immunophenotyping can be particularly useful in differential diagnosis between WM and related lymphomas (). The distinction between WM and splenic MZL is especially challenging. A WM clone will typically show homogenous CD25 expression with weak CD22, whereas patients with MZL will typically possess the opposite phenotype (CD25 negative with strong CD22), although variations can clearly occur in some patients. Other important differentiating markers include surface membrane IgM and CD79b, which are often over-expressed in WM, and CD305, which is upregulated in MZL but not in WM [Citation16,Citation41]. These phenotypic differences are demonstrable only by flow cytometry, suggesting that flow cytometry plus MYD88 analysis has more value than immunohistochemical assessment of trephine biopsies for differentiation of WM from splenic MZL.
5. Genotype
5.1. Cytogenetic abnormalities
The are no disease-defining cytogenetic abnormalities associated with WM, and a lack of highly recurrent translocations is also a distinguishing feature [Citation42,Citation43]. However, a range of cytogenetic abnormalities have been reported that appear to affect clinical features and outcomes. 6q deletion has been reported in 40–50% of WM cases and is associated with poor prognosis, as well as progression to symptomatic disease and a shorter time to progression to symptomatic disease [Citation44–46]. A copy number abnormality of 18q (+18q11-18q23) may also be common in WM and involved in malignant transformation [Citation45].
Conventional karyotyping is no longer conducted in WM diagnosis because of the difficulty in collecting samples of tumor cells in metaphase and without blood contamination. However, the use of fluorescence in situ hybridization may be considered to support a differential diagnosis. For example, t(11;14) is common in IgM multiple myeloma but absent in WM [Citation47,Citation48].
5.2. MYD88
The 5th edition of the WHO Classification of Haematolymphoid Tumours describes WM as a disease with two molecular subsets defined by the presence or absence of the MYD88 p.L265P (NP_002459.2) mutation [Citation3]. Several studies have reported MYD88L265P in >90% of patients with proven well-defined WM and other MYD88 mutations in ~4%, with only a small minority being wild-type for MYD88 [Citation46,Citation49–56]. Some studies from Japan and South Korea have reported a lower incidence of the MYD88L265P mutation [Citation57–59], but there is at this time no definitive evidence for significant differences according to ethnicity.
L265P and other activating mutations in MYD88 result in spontaneous assembly of protein complexes that lead to multiple pro-survival signaling cascades [Citation60]. Given its prevalence, mutational analysis of MYD88 is strongly recommended as part of the diagnostic procedure for WM [Citation18]. Testing for MYD88L265P can also be used to confirm WM involvement in complex extramedullary cases such as pleural effusions and Bing–Neel syndrome (LPL invasion of the leptomeningeal tissue and/or the central nervous system) [Citation61,Citation62].
It is also appropriate to consider whether patients with suspected WM and MYD88WT may have an alternative underlying pathology. In one study, 18 (28%) of 64 patients previously diagnosed with MYD88WT WM were considered to have an alternative clinicopathological diagnosis following review, with IgM multiple myeloma being the most common (n = 7, 11%) [Citation63]. IgM multiple myeloma may be distinguished from WM with careful assessment of all clinicopathological features. IgM multiple myeloma will be characterized by an infiltrate composed entirely of plasma cells and will lack monotypic B cells. The plasma cells will frequently express an aberrant or myeloma-like phenotype. WM by definition will be composed of both monotypic B cells and plasma cells. Genomic assessment has further utility, as IgM myeloma will frequently possess t(11;14) translocation, whereas MYD88L265P is typically absent [Citation48,Citation63]. Furthermore, IgM multiple myeloma patients will lack lymph node disease but may have lytic bone disease.
MYD88 mutational status has some prognostic implications for the clinical course and treatment response. MYD88WT is an independent risk factor for progression in asymptomatic WM patients [Citation64] and is associated with a greater risk of transformation than MYD88L265P [Citation63,Citation65,Citation66]. There are conflicting data regarding the impact of MYD88WT on overall survival; some studies demonstrate inferior survival outcomes compared with patients with the MYD88L265P mutation, but others do not [Citation63,Citation65–68]. However, it may be notable that Abeykoon et al., who did not detect a survival difference according to MYD88 status, reported a higher proportion of MYD88WT patients (21%) within their WM patient population than is typical [Citation65].
MYD88WT appears to be associated with a poorer response to the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib compared with MYD88L265P (), which has led some to propose genotype-based treatment strategies [Citation46]. However, the data to support such an approach are limited, and these algorithms may soon be outdated with the availability of next-generation BTK inhibitors. Higher response rates have been reported with acalabrutinib [Citation69] and zanubrutinib [Citation70] in MYD88WT WM patients. Indeed, zanubrutinib was prospectively evaluated in MYD88WT WM and is the first BTK inhibitor for which very good partial responses have been reported in this patient subset [Citation70] ().
Table 3. Patient responses to BTK inhibitors by MYD88 and CXCR4 status.
5.3. CXCR4
Activating mutations in the C-terminal domain of chemokine receptor gene CXCR4 are present in up to 40% of patients with WM who almost always also carry MYD88 mutations. CXCR4 mutations are rare in other lymphoproliferative diseases, with only sporadic cases reported in patients with MZL and the activated B-cell subtype of diffuse large B-cell lymphoma (DLBCL) [Citation46].
The mutational landscape of CXCR4 is complex, with >50 nonsense and frameshift mutations described. Interestingly, somatic CXCR4 mutations observed in WM tumor cells are similar to a germline mutation found in the immunodeficiency disorder warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome [Citation71]. Preclinical studies have shown that cells with the most common CXCR4S338X mutation are resistant to ibrutinib-triggered apoptosis and show sustained signaling of Ak strain transforming (AKT) and extracellular signal-regulating kinase (ERK) compared with wild-type CXCR4 cells [Citation72]. In contrast to MYD88L265P, CXCR4 mutations in WM are usually subclonal and a patient can harbor multiple mutations within separate clones [Citation46].
Mutational analysis of CXCR4 has limited utility in diagnosis and is not commonly performed in routine clinical practice as a consequence of this and the technical challenges in detection and interpretation. Beyond diagnosis, CXCR4 mutations are associated with increased bone marrow disease, serum IgM levels, symptomatic hyperviscosity, thrombocytopenia, and acquired von Willebrand factor deficiency at presentation [Citation67,Citation73]. CXCR4 mutations also appear to have prognostic utility in the context of BTK inhibitor–based therapy. Delayed and poorer responses to ibrutinib, as well as inferior progression-free survival, have been reported in patients with the MYD88L265P/CXCR4MUT genotype compared with MYD88L265P/CXCR4WT patients () [Citation74–77]. Long-term follow-up of the ASPEN head-to-head trial of zanubrutinib versus ibrutinib in WM indicates that the same is true for zanubrutinib, although there was a trend toward higher rates of complete and very good partial responses with zanubrutinib versus ibrutinib for MYD88L265P/CXCR4MUT patients (21% vs. 5%, P = 0.15) [Citation78].
There are limited data on the impact of CXCR4 in the context of other therapies [Citation18]. A study conducted by the French Innovative Leukemia Organization reported that CXCR4 mutational status had no impact on disease responses or survival outcomes of 69 patients treated with first-line bendamustine plus rituximab [Citation79]. Similarly, proteasome inhibitors, such as bortezomib and carfilzomib, appear to act independently of CXCR4 [Citation80,Citation81], while survival outcomes were not impacted in patients treated with idelalisib and obinutuzumab [Citation82].
5.4. Other somatic mutations
A range of somatic mutations in genes other than MYD88 or CXCR4 have been reported in patients with WM () [Citation83]. The number of detectable genetic abnormalities in IgM monoclonal gammopathies increases with the aggressiveness of the disease. In a study evaluating the 12 most frequently mutated genes in WM, the percentage of patients with alterations varied from 21% IgM MGUS, 35% asymptomatic WM, and 50% symptomatic WM [Citation84].
Figure 3. The frequency of somatic mutations occurring in WM (adapted from García-Sanz et al.) [Citation83].
![Figure 3. The frequency of somatic mutations occurring in WM (adapted from García-Sanz et al.) [Citation83].](/cms/asset/5d6cd949-8557-4cca-af32-c1cf7b5a09aa/ierr_a_2270779_f0003_oc.jpg)
TP53 aberrations, including mutations or deletion of the TP53 locus on chromosome 17 (17p13.1), have been observed in ~10% of patients with newly diagnosed WM and 25% at more advanced stages, and may be associated with mutated MYD88 and CXCR4 [Citation83,Citation85]. In common with other lymphomas, TP53 aberrations in WM appear to predict more aggressive disease [Citation85,Citation86]. While TP53 mutations predict for poor outcomes with immunochemotherapy in chronic lymphocytic leukemia [Citation87], definitive data in WM are lacking at this time.
Somatic mutations in ARID1A have been detected in 3–17% of patients with WM, including nonsense and frameshift variants, although no prognostic value has been demonstrated to date [Citation42,Citation56,Citation84]. ARID1A may modulate TP53 and is thought to act as an epigenetic tumor suppressor in ovarian cancer [Citation46]. In patients with WM, ARID1A mutations are associated with increased bone marrow infiltration [Citation88]; deletion of its homolog ARID1B (located on 6q) may contribute to the poor prognosis associated with 6q deletion.
Mutations in CD79A and CD79B can be found in ~10% of patients with WM [Citation46,Citation56,Citation61,Citation84,Citation91]. Both are components of the B-cell receptor (BCR) pathway and can form heterodimers with each other, so activating mutations of these components could contribute to the chronic BCR signaling observed in WM cells [Citation46]. In one study, mutations in CD79A and CD79B were found to be nearly exclusive of CXCR4 mutations, suggesting that CD79A/B mutations may have an independent role in facilitating mutated MYD88-directed progression in WM [Citation71]. In addition, CD79B mutations have been associated with histologic transformation in some WM patients [Citation92]. An association of CD79B and MYD88 mutations has also been described in activated B-cell-like DLBCL [Citation60].
6. Methodologies for MYD88 and CXCR4 mutational analysis
There are no standardized protocols for mutational analysis of MYD88 or CXCR4 in patients with WM (). DNA for mutational analysis of MYD88 or CXCR4 can be extracted from bone marrow, peripheral blood, or plasma [Citation18,Citation21]. However, the use of peripheral blood can give false-negatives and is not recommended in routine diagnosis [Citation18]. Several studies indicate that mutational analysis of cell-free DNA from plasma can be reliably used to identify MYD88L265P and CXCR4S338X [Citation93–95]. However, further confirmation is needed before this approach can be recommended in clinical practice.
Table 4. Key genes and methodologies in WM diagnosis and prognosis.
Allele-specific (AS) polymerase chain reaction (PCR) is used by most laboratories to detect MYD88L265P because it is relatively simple and highly sensitive. More recently, droplet digital PCR (ddPCR) has emerged as an alternative that allows absolute quantification of DNA and provides higher sensitivity than AS-PCR [Citation96,Citation97]. Drandi et al. validated the use of ddPCR for the detection of MYD88L265P and reported a sensitivity of 5.0 × 10−5 for ddPCR versus 1.0 × 10−3 for AS-PCR [Citation96]. The sensitivity offered by ddPCR will reduce the risk of false-negative assessments and has clear utility in the assessment of patients with low-level marrow disease, such as IgM MGUS and IgM-related disorders. The sensitivity of ddPCR might also allow for peripheral blood diagnosis in elderly and unfit patients.
Drandi et al. explored the potential for MYD88L265P mutation detection by ddPCR to be used for minimal residual disease (MRD) monitoring, finding that the assay correlated with an IGH-based MRD ddPCR assay, which is widely used but not validated for WM [Citation96]. In this small study, 6/22 (27%) patients with WM were MRD-negative (MYD88L265P <5.0 × 10−5) in the bone marrow and 17/35 (49%) in the peripheral blood after first-line therapy. Fludarabine- and bendamustine-containing regimens showed a high potential for MRD negativity, and MRD monitoring could potentially be used to determine the duration of therapy and whether maintenance therapy is required following treatment [Citation96]. Such MRD-based strategies may provide a more detailed evaluation of response and survival outcomes given the low incidence of complete responses, particularly in the context of conventional rituximab-based therapies. It is, however, important to be aware that MYD88L265P has been detected in healthy mature B cells [Citation98].
While MYD88 profiling can be performed on whole bone marrow samples, CD19+ cell selection using magnetic beads or flow sorting is recommended for detection of subclonal mutations (e.g. CXCR4 and TP53). Ideally, these mutations should be evaluated by next-generation sequencing (NGS) approaches, but targeted sequencing could have a role in mutational analysis of CXCR4. Patients with WM may have CXCR4 frameshift or nonsense mutations, but only nonsense mutations have been associated with an increased risk of progression and/or death versus CXCR4WT, and 80–90% of CXCR4 nonsense mutations in WM are located in S338X [Citation99,Citation100]. Therefore, an AS-PCR or ddPCR assay targeting CXCR4S338X may be sufficient in a routine diagnostic setting.
In patients who are negative for MYD88L265P where a diagnosis of WM is strongly suspected, Sanger sequencing or, ideally, NGS may be used to identify other MYD88 mutations or mutations associated with other lymphoproliferative syndromes, such as KLF2 or NOTCH2 in MZL [Citation101]. CD19+ selection is again recommended in this setting [Citation46].
If CD19+ selection is not feasible, it is important that laboratories are aware that there is a high risk of false-negative results with targeted NGS of MYD88 or CXCR4, particularly when a sample is contaminated with blood. A detection limit of 1% is required when using NGS to detect MYD88 or CXCR4 variants. In a study comparing the sensitivity of a clinically validated and targeted NGS assay (Rapid Heme Panel) with AS-PCR, the targeted NGS of unselected bone marrow samples was associated with 69% sensitivity for MYD88L265P and 16% sensitivity for CXCR4S338X compared with AS-PCR [Citation102].
7. Conclusion
Accurate and timely diagnosis of WM is essential to ensure appropriate treatment, optimize outcomes, and preserve quality of life for patients. The heterogeneity of patient presentations in WM poses a particular challenge in diagnosis, but advances in clinical understanding and technology in the past decade have provided a much stronger basis for pathological diagnosis. The identification of MYD88L265P as a differentiating feature of WM is particularly useful, but in the absence of standardized testing protocols it is essential that laboratories adopt appropriate assays and determine their sensitivities.
Genotyping in WM outside of clinical trials is usually limited to MYD88, but data suggest that CXCR4 and TP53 mutations can support prognosis and treatment decisions. More research is needed to determine the value of these genes to diagnosis, prognosis, and treatment decisions.
8. Expert opinion
WM is defined by the presence of morphologically detectable LPL on an adequate trephine biopsy in the context of IgM monoclonal gammopathy [Citation1–3]. The use of arbitrary levels of bone marrow disease for disease definition are inappropriate, given that symptomatic disease requiring therapy can occur in patients with less than 10% infiltration [Citation25].
Diagnostic guidelines for WM are consistent regarding the need for a bone marrow biopsy to achieve a definitive diagnosis [Citation18,Citation103]. A biopsy is essential to establish LPL in the bone marrow and quantify the degree of infiltration. However, accurate noninvasive alternative paths to diagnosis are highly desirable in a disease of elderly patients. Immunophenotyping is a powerful diagnostic tool that has the potential to provide the basis for such an approach. The accuracy of diagnosing WM based on flow cytometry immunophenotyping combined with MYD88 testing from bone marrow aspirates should be evaluated compared with bone marrow biopsy–based diagnosis. However, there are significant barriers to such an approach. Identifying the hallmark WM immunophenotype is difficult and requires considerable technical expertise. As such, flow cytometry is more likely to be used by most centers to establish the presence of clonal B cells and to differentiate between related lymphoid diseases, such as chronic lymphocytic leukemia.
The recent WHO classifications of WM describe two molecular subsets based on the presence or absence of MYD88 p.L265P [Citation3]; it would be more accurate to define molecular subtypes based on the presence or absence of all activating MYD88 mutations and not exclusively L265P. Studies indicate that patients with WM and wild-type MYD88 have a higher risk of transformation, shorter overall survival, and respond poorly to certain therapies compared with patients with mutated MYD88 [Citation63–68]. However, the interpretation of the data is complicated by a lack of standardization in the methodologies used to test for MYD88 mutations across trials. A comparison of outcomes for patients without MYD88 mutations from the iNNOVATE trial of ibrutinib plus rituximab versus rituximab monotherapy and the pivotal ibrutinib study is illustrative.
MYD88WT patients in the iNNOVATE trial treated with ibrutinib plus rituximab had much higher major response rates (73% vs. 0%) and much longer progression-free survival (>4 vs. 0.4 years) compared with MYD88WT patients treated with ibrutinib monotherapy in the pivotal study. However, the iNNOVATE trial used an NGS assay with either unselected bone marrow aspirate or formalin-fixed paraffin-embedded specimens to identify MYD88 mutations, whereas the pivotal ibrutinib trial used AS-PCR and Sanger sequencing with CD19-selected bone marrow aspirate. The relative percentage of MYD88WT patients in the iNNOVATE trial was much higher than in the ibrutinib monotherapy study (16% vs. 6%) and likely reflects the lower sensitivity of the methodology and the presence of patients misclassified as MYD88WT [Citation74,Citation75,Citation104–106].
Related to the need for standardization, it is essential to report the sensitivity and specificity of assays used to test for MYD88 status. These parameters must also be defined outside of clinical trials. MYD88 status can determine the differential diagnosis of WM from related conditions and the potential for false-negatives must be considered, as well as the potential for activating MYD88 mutations other than L265P.
The importance of an accurate assessment of MYD88 status was repeatedly stressed during the 11th International Workshop on Waldenström’s Macroglobulinemia (IWWM). The panel recommended the adoption of ddPCR as the gold standard for MYD88 testing. The higher sensitivity of ddPCR compared with AS-PCR reduces the risk of false-negatives and misdiagnosis [Citation96]. Its higher sensitivity also increases the potential to test for mutations using non–bone marrow sources of DNA, such as cell-free DNA [Citation96]. Noninvasive approaches to molecular testing will be particularly important if/when MRD monitoring is incorporated into post-treatment evaluation in WM.
More research is needed to determine the value of evaluating the mutational status of CXCR4, TP53, and other genes that are mutated at a lesser frequency than MYD88. Routine analysis of CXCR4 is difficult and costly, but CXCR4 mutations are considered essentially unique to WM and impact response rates and survival outcomes with BTK inhibitors [Citation46]. Accordingly, measures to include CXCR4 mutations in diagnosis should be investigated. Accurate detection of CXCR4 mutations will require NGS coupled with CD19+ cell selection. Similarly, TP53 requires further study in WM using similar approaches.
Molecular-based treatment algorithms have been proposed for WM, and we would encourage detailed genomic assessments in all clinical trials so that these can be further refined and applied to routine clinical management.
Article highlights
Waldenström’s macroglobulinemia (WM) is an indolent B-cell lymphoplasmacytic lymphoma (LPL) with immunoglobulin M (IgM) monoclonal gammopathy and morphologic evidence of bone marrow infiltration by LPL.
About a quarter of patients with WM are asymptomatic at the time of diagnosis; anemia is the primary reason for patients with WM to present to primary care.
Bone marrow biopsy is essential for a definitive diagnosis of WM.
A typical WM immunophenotype is a powerful and potentially underutilized tool in the differential diagnosis of WM from related conditions, such as marginal zone lymphoma.
The WM clone expresses pan-B-cell markers (CD19, CD20, CD22, Sig) but can be distinguished from normal B cells by flow cytometry based on weak CD22 expression and homogeneous positivity for CD25.
The MYD88 p.L265P (NP_002459.2) mutation is found in >90% of patients with WM and is strongly recommended in the diagnosis of patients with suspected WM.
CXCR4 and TP53, which are mutated at a lesser frequency than MYD88, have limited relevance in establishing a diagnosis of WM but have some prognostic value.
There are no standardized protocols for mutational analysis of MYD88; allele-specific (AS) polymerase chain reaction (PCR) is used by most laboratories to detect MYD88L265P, but droplet digital PCR has emerged as an alternative that allows absolute quantification of DNA and provides higher sensitivity than AS-PCR.
Declaration of interest
RGS declares honoraria from Astellas, BeiGene, Incyte, Janssen, Novartis, the Spanish Public National System, and Takeda; consulting services to BeiGene and Takeda; and research funding from Astellas, Gilead, Incyte, IVS Technologies (patent partial use), Janssen, the Spanish Society of Hematology and Hemotherapy, and Takeda.
RO has received honoraria from Janssen, BeiGene, Celgene, and AstraZeneca; and has provided advisory services to BeiGene and Janssen.
MV has received honoraria from AbbVie, AstraZeneca, BeiGene, and Janssen; and provided advisory services to AbbVie, AstraZeneca, BeiGene, and Janssen.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Acknowledgments
Editorial support was provided by Luke Smith, PhD, of Porterhouse Medical, to collate and incorporate author input on the manuscript, all carried out under the authors’ direction.
Additional information
Funding
References
- Campo E, Swerdlow SH, Harris NL, et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. doi: 10.1182/blood-2011-01-293050
- Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003;30(2):110–115. doi: 10.1053/sonc.2003.50082
- Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720–1748. doi: 10.1038/s41375-022-01620-2
- Wang H, Chen Y, Li F, et al. Temporal and geographic variations of Waldenstrom macroglobulinemia incidence: a large population-based study. Cancer. 2012;118(15):3793–3800. doi: 10.1002/cncr.26627
- Groves FD, Travis LB, Devesa SS, et al. Waldenström’s macroglobulinemia: incidence patterns in the United States, 1988-1994. Cancer. 1998;82(6):1078–1081. doi: 10.1002/(SICI)1097-0142(19980315)82:6<1078::AID-CNCR10>3.0.CO;2-3
- Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443–459. doi: 10.3322/caac.21357
- Buske C, Leblond V. How to manage Waldenstrom’s macroglobulinemia. Leukemia. 2013;27(4):762–772. doi: 10.1038/leu.2013.36
- de Tute RM, Rawstron AC, Owen RG. Immunoglobulin M concentration in Waldenström macroglobulinemia: correlation with bone marrow B cells and plasma cells. Clin Lymphoma Myeloma Leuk. 2013;13(2):211–213. doi: 10.1016/j.clml.2013.02.018
- Pasricha S-R, Juneja SK, Westerman DA, et al. Bone-marrow plasma cell burden correlates with IgM paraprotein concentration in Waldenström macroglobulinaemia. J Clin Pathol. 2011;64(6):520–523. doi: 10.1136/jcp.2010.088591
- Menke MN, Feke GT, McMeel JW, et al. Ophthalmologic techniques to assess the severity of hyperviscosity syndrome and the effect of plasmapheresis in patients with Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma. 2009;9(1):100–103. doi: 10.3816/CLM.2009.n.027
- Olszewski AJ, Treon SP, Castillo JJ. Evolution of management and outcomes in Waldenström macroglobulinemia: a population-based analysis. Oncologist. 2016;21(11):1377–1386. doi: 10.1634/theoncologist.2016-0126
- Kristinsson SY, Eloranta S, Dickman PW, et al. Patterns of survival in lymphoplasmacytic lymphoma/Waldenström macroglobulinemia: a population-based study of 1,555 patients diagnosed in Sweden from 1980 to 2005. Am J Hematol. 2013;88(1):60–65. doi: 10.1002/ajh.23351
- Amaador K, Kersten MJ, Visser O, et al. Primary therapy and relative survival in patients with lymphoplasmacytic lymphoma/Waldenström macroglobulinaemia: a population-based study in the Netherlands, 1989–2018. Br J Haematol. 2022;196(3):660–669. doi: 10.1111/bjh.17856
- Chien H-C, Morreall D, Patil V, et al. Real-world treatment patterns and outcomes in a national study of veterans with Waldenström macroglobulinemia, 2006–2019. Am J Hematol. 2021;96(6):E184–E187. doi: 10.1002/ajh.26133
- García-Sanz R, Montoto S, Torrequebrada A, et al. Waldenström macroglobulinaemia: presenting features and outcome in a series with 217 cases. Br J Haematol. 2001;115(3):575–582. doi: 10.1046/j.1365-2141.2001.03144.x
- Dimopoulos MA, Kastritis E. How I treat Waldenström macroglobulinemia. Blood. 2019;134(23):2022–2035. doi: 10.1182/blood.2019000725
- Vos JM, Gustine J, Rennke HG, et al. Renal disease related to Waldenström macroglobulinaemia: incidence, pathology and clinical outcomes. Br J Haematol. 2016;175(4):623–630. doi: 10.1111/bjh.14279
- Kastritis E, Leblond V, Dimopoulos MA, et al. Waldenström’s macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv41–iv50. doi: 10.1093/annonc/mdy146
- Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenström macroglobulinemia. Blood. 2009;113(18):4163–4170. doi: 10.1182/blood-2008-08-174961
- Owen RG, Kyle RA, Stone MJ, et al. Response assessment in Waldenström macroglobulinaemia: update from the VIth International Workshop. Br J Haematol. 2013;160(2):171–176. doi: 10.1111/bjh.12102
- Castillo JJ, Garcia-Sanz R, Hatjiharissi E, et al. Recommendations for the diagnosis and initial evaluation of patients with Waldenström macroglobulinaemia: a task force from the 8th International Workshop on Waldenström Macroglobulinaemia. Br J Haematol. 2016;175(1):77–86. doi: 10.1111/bjh.14196
- Morel P, Monconduit M, Jacomy D, et al. Prognostic factors in Waldenström macroglobulinemia: a report on 232 patients with the description of a new scoring system and its validation on 253 other patients. Blood. 2000;96(3):852–858 doi:10.1182/blood.V96.3.852.
- Leleu X, Xie W, Bagshaw M, et al. The role of serum immunoglobulin free light chain in response and progression in Waldenstrom macroglobulinemia. Clin Cancer Res. 2011;17(9):3013–3018. doi: 10.1158/1078-0432.CCR-10-2954
- Greil C, Ihorst G, Gaiser F, et al. The serum heavy/light chain immunoassay: a valuable tool for sensitive paraprotein assessment, risk, and disease monitoring in monoclonal gammopathies. Eur J Haematol. 2017;99(5):449–458. doi: 10.1111/ejh.12958
- Treon SP, Tedeschi A, San-Miguel J, et al. Report of consensus panel 4 from the 11th International Workshop on Waldenstrom’s Macroglobulinemia on diagnostic and response criteria. Semin Hematol. 2023;60(2):97–106. doi: 10.1053/j.seminhematol.2023.03.009
- Pan Q, Cao X, Luo Y, et al. Baseline 18F-FDG PET/CT may portend the prognosis of patients with Waldenström macroglobulinemia/lymphoplasmacytic lymphoma after first-line treatment. Clin Nucl Med. 2022;47(11):954–960. doi: 10.1097/RLU.0000000000004362
- Banwait R, O’Regan K, Campigotto F, et al. The role of 18F-FDG PET/CT imaging in Waldenstrom macroglobulinemia. Am J Hematol. 2011;86(7):567–572. doi: 10.1002/ajh.22044
- Kyle RA, Benson J, Larson D, et al. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma. 2009;9(1):17–18. doi: 10.3816/CLM.2009.n.002
- Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenström macroglobulinemia: long-term results. Blood. 2012;119(19):4462–4466. doi: 10.1182/blood-2011-10-384768
- Varettoni M, Zibellini S, Boveri E, et al. A risk-stratification model based on the initial concentration of the serum monoclonal protein and MYD88 mutation status identifies a subset of patients with IgM monoclonal gammopathy of undetermined significance at high risk of progression to Waldenström macroglobulinaemia or other lymphoproliferative disorders. Br J Haematol. 2019;187(4):441–446. doi: 10.1111/bjh.16086
- Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018;378(3):241–249. doi: 10.1056/NEJMoa1709974
- Garcia-Reyero J, Martinez Magunacelaya N, Gonzalez de Villambrosia S, et al. Diagnostic value of bone marrow core biopsy patterns in lymphoplasmacytic lymphoma/Waldenström macroglobulinaemia and description of its mutational profiles by targeted NGS. J Clin Pathol. 2020;73(9):571–577. doi: 10.1136/jclinpath-2019-206282
- Lemal R, Poulain S, Ledoux-Pilon A, et al. Mast cell density and its clinical relevance in Waldenström’s macroglobulinemia. EJHaem. 2022;3(2):371–378. doi: 10.1002/jha2.378
- Bassarova A, Trøen G, Spetalen S, et al. Lymphoplasmacytic lymphoma and marginal zone lymphoma in the bone marrow: paratrabecular involvement as an important distinguishing feature. Am J Clin Pathol. 2015;143(6):797–806. doi: 10.1309/AJCP6ZODWV1CIDME
- Amaador K, Vos JMI, Pals ST, et al. Discriminating between Waldenström macroglobulinemia and marginal zone lymphoma using logistic LASSO regression. Leuk Lymphoma. 2022;63(5):1070–1079. doi: 10.1080/10428194.2021.2018584
- Maqbool MG, Tam CS, Morison IM, et al. A practical guide to laboratory investigations at diagnosis and follow up in Waldenström macroglobulinaemia: recommendations from the Medical and Scientific Advisory Group, Myeloma Australia, the Pathology Sub-committee of the Lymphoma and Related Diseases Registry and the Australasian Association of Clinical Biochemists Monoclonal Gammopathy Working Group. Pathology. 2020;52(2):167–178. doi: 10.1016/j.pathol.2019.11.002
- Paiva B, Montes MC, García-Sanz R, et al. Multiparameter flow cytometry for the identification of the Waldenström’s clone in IgM-MGUS and Waldenström’s macroglobulinemia: new criteria for differential diagnosis and risk stratification. Leukemia. 2014;28(1):166–173. doi: 10.1038/leu.2013.124
- San Miguel JF, Vidriales MB, Ocio E, et al. Immunophenotypic analysis of Waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30(2):187–195. doi: 10.1053/sonc.2003.50074
- González D, van der Burg M, García-Sanz R, et al. Immunoglobulin gene rearrangements and the pathogenesis of multiple myeloma. Blood. 2007;110(9):3112–3121. doi: 10.1182/blood-2007-02-069625
- Corbingi A, Innocenti I, Tomasso A, et al. Monoclonal gammopathy and serum immunoglobulin levels as prognostic factors in chronic lymphocytic leukaemia. Br J Haematol. 2020;190(6):901–908. doi: 10.1111/bjh.16975
- Jelinek T, Bezdekova R, Zatopkova M, et al. Current applications of multiparameter flow cytometry in plasma cell disorders. Blood Cancer J. 2017;7(10):e617. doi: 10.1038/bcj.2017.90
- Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637–1646. doi: 10.1182/blood-2013-09-525808
- Nguyen-Khac F, Lambert J, Chapiro E, et al. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenström’s macroglobulinemia. Haematologica. 2013;98(4):649–654. doi: 10.3324/haematol.2012.070458
- García-Sanz R, Dogliotti I, Zaccaria GM, et al. 6q deletion in Waldenström macroglobulinaemia negatively affects time to transformation and survival. Br J Haematol. 2021;192(5):843–852. doi: 10.1111/bjh.17028
- Paiva B, Corchete LA, Vidriales MB, et al. The cellular origin and malignant transformation of Waldenström macroglobulinemia. Blood. 2015;125(15):2370–2380. doi: 10.1182/blood-2014-09-602565
- Treon SP, Xu L, Guerrera ML, et al. Genomic landscape of Waldenström macroglobulinemia and its impact on treatment strategies. J Clin Oncol. 2020;38(11):1198–1208. doi: 10.1200/JCO.19.02314
- Askari E, Rodriguez S, Garcia-Sanz R. Waldenström’s macroglobulinemia: an exploration into the pathology and diagnosis of a complex B-cell malignancy. J Blood Med. 2021;12:795–807. doi: 10.2147/JBM.S267938
- Bazarbachi AH, Avet-Loiseau H, Szalat R, et al. IgM-MM is predominantly a pre–germinal center disorder and has a distinct genomic and transcriptomic signature from WM. Blood. 2021;138(20):1980–1985. doi: 10.1182/blood.2021011452
- Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367(9):826–833. doi: 10.1056/NEJMoa1200710
- Jiménez C, Sebastián E, Chillón MC, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenström’s macroglobulinemia. Leukemia. 2013;27(8):1722–1728. doi: 10.1038/leu.2013.62
- Poulain S, Roumier C, Decambron A, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121(22):4504–4511. doi: 10.1182/blood-2012-06-436329
- Varettoni M, Arcaini L, Zibellini S, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenström’s macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121(13):2522–2528. doi: 10.1182/blood-2012-09-457101
- Xu L, Hunter ZR, Yang G, et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013;121(11):2051–2058. doi: 10.1182/blood-2012-09-454355
- Gustine J, Meid K, Xu L, et al. To select or not to select? The role of B-cell selection in determining the MYD88 mutation status in Waldenström macroglobulinaemia. Br J Haematol. 2017;176(5):822–824. doi: 10.1111/bjh.13996
- Treon SP, Xu L, Hunter Z. MYD88 mutations and response to ibrutinib in Waldenström’s macroglobulinemia. N Engl J Med. 2015;373(6):584–586. doi: 10.1056/NEJMc1506192
- Varettoni M, Zibellini S, Defrancesco I, et al. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. 2017;102(12):2077–2085. doi: 10.3324/haematol.2017.172718
- Nakamura A, Ohwada C, Takeuchi M, et al. Detection of MYD88 L265P mutation by next-generation deep sequencing in peripheral blood mononuclear cells of Waldenström’s macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. PLoS One. 2019;14(9):e0221941. doi: 10.1371/journal.pone.0221941
- Mori N, Ohwashi M, Yoshinaga K, et al. L265P mutation of the MYD88 gene is frequent in Waldenström’s macroglobulinemia and its absence in myeloma. PLoS One. 2013;8(11):e80088. doi: 10.1371/journal.pone.0080088
- Kim J-A, Im K, Park SN, et al. MYD88 L265P mutations are correlated with 6q deletion in Korean patients with Waldenström macroglobulinemia. Biomed Res Int. 2014;2014:363540. doi: 10.1155/2014/363540
- Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. doi: 10.1038/nature09671
- Gustine JN, Meid K, Hunter ZR, et al. MYD88 mutations can be used to identify malignant pleural effusions in Waldenström macroglobulinaemia. Br J Haematol. 2018;180(4):578–581. doi: 10.1111/bjh.14386
- Poulain S, Boyle EM, Roumier C, et al. MYD88 L265P mutation contributes to the diagnosis of Bing Neel syndrome. Br J Haematol. 2014;167(4):506–513. doi: 10.1111/bjh.13078
- Treon SP, Gustine J, Xu L, et al. MYD88 wild-type Waldenstrom macroglobulinaemia: differential diagnosis, risk of histological transformation, and overall survival. Br J Haematol. 2018;180(3):374–380. doi: 10.1111/bjh.15049
- Bustoros M, Sklavenitis-Pistofidis R, Kapoor P, et al. Progression risk stratification of asymptomatic Waldenström macroglobulinemia. J Clin Oncol. 2019;37(16):1403–1411. doi: 10.1200/JCO.19.00394
- Abeykoon JP, Paludo J, King RL, et al. MYD88 mutation status does not impact overall survival in Waldenström macroglobulinemia. Am J Hematol. 2018;93(2):187–194. doi: 10.1002/ajh.24955
- Zanwar S, Abeykoon JP, Durot E, et al. Impact of MYD88L265P mutation status on histological transformation of Waldenström macroglobulinemia. Am J Hematol. 2020;95(3):274–281. doi: 10.1002/ajh.25697
- Treon SP, Cao Y, Xu L, et al. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenström macroglobulinemia. Blood. 2014;123(18):2791–2796. doi: 10.1182/blood-2014-01-550905
- Wang Y, Gali VL, Xu-Monette ZY, et al. Molecular and genetic biomarkers implemented from next-generation sequencing provide treatment insights in clinical practice for Waldenström macroglobulinemia. Neoplasia. 2021;23(4):361–374. doi: 10.1016/j.neo.2021.02.002
- Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: a single-arm, multicentre, phase 2 study. Lancet Haematol. 2020;7(2):e112–e121. doi: 10.1016/S2352-3026(19)30210-8
- Dimopoulos M, Garcia-Sanz R, Lee HP, et al. Zanubrutinib for the treatment of MYD88 wild-type Waldenström macroglobulinemia: a substudy of the phase 3 ASPEN trial. Blood Adv. 2020;4(23):6009–6018. doi: 10.1182/bloodadvances.2020003010
- Poulain S, Roumier C, Venet-Caillault A, et al. Genomic landscape of CXCR4 mutations in Waldenström macroglobulinemia. Clin Cancer Res. 2016;22(6):1480–1488. doi: 10.1158/1078-0432.CCR-15-0646
- Cao Y, Hunter ZR, Liu X, et al. The WHIM-like CXCR4S338X somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s macroglobulinemia. Leukemia. 2015;29(1):169–176. doi: 10.1038/leu.2014.187
- Castillo JJ, Gustine JN, Meid K, et al. Low levels of von Willebrand markers associate with high serum IgM levels and improve with response to therapy, in patients with Waldenström macroglobulinaemia. Br J Haematol. 2019;184(6):1011–1014. doi: 10.1111/bjh.15200
- Treon SP, Meid K, Gustine J, et al. Long-term follow-up of ibrutinib monotherapy in symptomatic, previously treated patients with Waldenström macroglobulinemia. J Clin Oncol. 2021;39(6):565–575. doi: 10.1200/JCO.20.00555
- Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N Engl J Med. 2015;372(15):1430–1440. doi: 10.1056/NEJMoa1501548
- Dimopoulos MA, Trotman J, Tedeschi A, et al. Ibrutinib for patients with rituximab-refractory Waldenström’s macroglobulinaemia (iNNOVATE): an open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017;18(2):241–250. doi: 10.1016/S1470-2045(16)30632-5
- Treon SP, Gustine J, Meid K, et al. Ibrutinib monotherapy in symptomatic, treatment-naïve patients with Waldenström macroglobulinemia. J Clin Oncol. 2018;36(27):2755–2761. doi: 10.1200/JCO.2018.78.6426
- Tam CSL, Garcia-Sanz R, Opat S, et al. ASPEN: long-term follow-up results of a phase 3 randomized trial of zanubrutinib (ZANU) versus ibrutinib (IBR) in patients with Waldenström macroglobulinemia (WM). J Clin Oncol. 2022;40(16 suppl):7521. doi: 10.1200/JCO.2022.40.16_suppl.7521
- Laribi K, Poulain S, Willems L, et al. Bendamustine plus rituximab in newly-diagnosed Waldenström macroglobulinaemia patients. A study on behalf of the French Innovative Leukaemia Organization (FILO). Br J Haematol. 2019;186(1):146–149. doi: 10.1111/bjh.15718
- Sklavenitis-Pistofidis R, Capelletti M, Liu CJ, et al. Bortezomib overcomes the negative impact of CXCR4 mutations on survival of Waldenstrom macroglobulinemia patients. Blood. 2018;132(24):2608–2612. doi: 10.1182/blood-2018-07-863241
- Treon SP, Tripsas CK, Meid K, et al. Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenström’s macroglobulinemia. Blood. 2014;124(4):503–510. doi: 10.1182/blood-2014-03-566273
- Tomowiak C, Poulain S, Herbaux C, et al. Obinutuzumab and idelalisib in symptomatic patients with relapsed/refractory Waldenström macroglobulinemia. Blood Adv. 2021;5(9):2438–2446. doi: 10.1182/bloodadvances.2020003895
- García-Sanz R, Jiménez C. Time to move to the single-cell level: applications of single-cell multi-omics to hematological malignancies and Waldenström’s macroglobulinemia—a particularly heterogeneous lymphoma. Cancers (Basel). 2021;13(7):1541. doi: 10.3390/cancers13071541.
- Jiménez C, Prieto-Conde MI, García-Álvarez M, et al. Unraveling the heterogeneity of IgM monoclonal gammopathies: a gene mutational and gene expression study. Ann Hematol. 2018;97(3):475–484. doi: 10.1007/s00277-017-3207-3
- Gustine JN, Tsakmaklis N, Demos MG, et al. TP53 mutations are associated with mutated MYD88 and CXCR4, and confer an adverse outcome in Waldenström macroglobulinaemia. Br J Haematol. 2019;184(2):242–245. doi: 10.1111/bjh.15560
- Poulain S, Roumier C, Bertrand E, et al. TP53 mutation and its prognostic significance in Waldenstrom’s macroglobulinemia. Clin Cancer Res. 2017;23(20):6325–6335. doi: 10.1158/1078-0432.CCR-17-0007
- Eichhorst B, Robak T, Montserrat E, et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(1):23–33. doi: 10.1016/j.annonc.2020.09.019
- Hunter ZR, Xu L, Yang G, et al. Transcriptome sequencing reveals a profile that corresponds to genomic variants in Waldenström macroglobulinemia. Blood. 2016;128(6):827–838. doi: 10.1182/blood-2016-03-708263
- Trotman J, Opat S, Gottlieb D, et al. Zanubrutinib for the treatment of patients with Waldenström macroglobulinemia: 3 years of follow-up. Blood. 2020;136(18):2027–2037. doi: 10.1182/blood.2020006449
- Tam CS, Opat S, D’Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–2050. doi: 10.1182/blood.2020006844
- Poulain S, Roumier C, Galiègue-Zouitina S, et al. Genome wide SNP array identified multiple mechanisms of genetic changes in Waldenstrom macroglobulinemia. Am J Hematol. 2013;88(11):948–954. doi: 10.1002/ajh.23545
- Jiménez C, Alonso-Álvarez S, Alcoceba M, et al. From Waldenström’s macroglobulinemia to aggressive diffuse large B-cell lymphoma: a whole-exome analysis of abnormalities leading to transformation. Blood Cancer J. 2017;7(8):e591. doi: 10.1038/bcj.2017.72
- Ntanasis-Stathopoulos I, Bagratuni T, Gavriatopoulou M, et al. Cell-free DNA analysis for the detection of MYD88 and CXCR4 mutations in IgM monoclonal gammopathies; an update with clinicopathological correlations. Am J Hematol. 2020;95(6):E148–E150. doi: 10.1002/ajh.25802
- Wu Y-Y, Jia M-N, Cai H, et al. Detection of the MYD88L265P and CXCR4S338X mutations by cell-free DNA in Waldenström macroglobulinemia. Ann Hematol. 2020;99(8):1763–1769. doi: 10.1007/s00277-020-04139-7
- Demos MG, Hunter ZR, Xu L, et al. Cell-free DNA analysis for detection of MYD88L265P and CXCR4S338X mutations in Waldenström macroglobulinemia. Am J Hematol. 2021;96(7):E250–E253. doi: 10.1002/ajh.26184
- Drandi D, Genuardi E, Dogliotti I, et al. Highly sensitive MYD88L265P mutation detection by droplet digital polymerase chain reaction in Waldenström macroglobulinemia. Haematologica. 2018;103(6):1029–1037. doi: 10.3324/haematol.2017.186528
- Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–8610. doi: 10.1021/ac202028g
- Rodriguez S, Celay J, Goicoechea I, et al. Preneoplastic somatic mutations including MYD88L265P in lymphoplasmacytic lymphoma. Sci Adv. 2022;8(3):eabl4644. doi: 10.1126/sciadv.abl4644
- Castillo JJ, Xu L, Gustine JN, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br J Haematol. 2019;187(3):356–363. doi: 10.1111/bjh.16088
- Kaiser LM, Hunter ZR, Treon SP, et al. CXCR4 in Waldenström’s macroglobulinema: chances and challenges. Leukemia. 2021;35(2):333–345. doi: 10.1038/s41375-020-01102-3
- Campos-Martín Y, Martínez N, Martínez-López A, et al. Clinical and diagnostic relevance of NOTCH2-and KLF2-mutations in splenic marginal zone lymphoma. Haematologica. 2017;102(8):e310–e312. doi: 10.3324/haematol.2016.161711
- Gustine JN, Xu L, Yang G, et al. Bone marrow involvement and subclonal diversity impairs detection of mutated CXCR4S338X by diagnostic next-generation sequencing in Waldenström macroglobulinaemia. Br J Haematol. 2021;194(4):730–733. doi: 10.1111/bjh.17385
- Garcia-Sanz R, Varettoni M, Jiménez C, et al. Report of consensus panel 3 from the 11th International Workshop on Waldenström’s Macroglobulinemia: recommendations for molecular diagnosis in Waldenström’s macroglobulinemia. Semin Hematol. 2023;60(2):90–96. doi: 10.1053/j.seminhematol.2023.03.007.
- Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 trial of ibrutinib plus rituximab in Waldenström’s macroglobulinemia. N Engl J Med. 2018;378(25):2399–2410. doi: 10.1056/NEJMoa1802917
- Buske C, Tedeschi A, Trotman J, et al. Ibrutinib plus rituximab versus placebo plus rituximab for Waldenström’s macroglobulinemia: final analysis from the randomized phase III iNNOVATE study. J Clin Oncol. 2022;40(1):52–62. doi: 10.1200/JCO.21.00838
- Kofides A, Hunter ZR, Xu L, et al. Diagnostic next-generation sequencing frequently fails to detect MYD88L265P in Waldenström macroglobulinemia. Hemasphere. 2021;5(8):e624. doi: 10.1097/HS9.0000000000000624