
Abstract
Purpose
To interpret the Medical Device Regulation for occupational therapists in the Netherlands involved in prescribing and manufacturing custom-made assistive devices and to develop a roadmap for implementation.
Materials and methods
Four online iterative co-design workshops were organized under supervision of a senior quality manager to assist in the interpretation of the MDR framework with a focus on custom-made assistive devices; and to assist the implementation by generating guidelines and forms. The workshops for seven participating occupational therapists had an interactive character with Q&A, small and homework assignments, and oral evaluations. Next to occupational therapists, participants with different backgrounds joined such as 3D printing experts, engineers, managers, and researchers.
Results
The participants experienced the interpretation of the MDR as informative, but also as complex. Complying with the MDR requires considerable documentation activities that are currently not part of care professionals tasks. This initially raised concerns regarding implementation in daily practice. To facilitate the MDR implementation, forms were created and evaluated for a selected design case together with the participants for future reference. Additionally, instructions were given which forms should be filled out only once per organization, which forms could be reused for similar types of custom-made devices, and which forms should be filled out for each individual custom-made device.
Conclusions
This study provides practical guidelines and forms to support occupational therapists in the Netherlands to prescribe and manufacture custom-made medical devices complying with the MDR. It is recommended to involve engineers and/or quality managers in this process.
IMPLICATIONS FOR REHABILITATION
Occupational therapists are considered legal manufacturer when they prescribe and manufacture custom-made medical devices for their clients. As such they are legally obliged to meet the Medical Device Regulation (MDR).
When designing and manufacturing “in-house” custom-made medical devices, care organizations need to follow and document activities to demonstrate compliance with the MDR. This study offers practical guidelines and forms to facilitate this.
Introduction
All products and devices launched onto the market or into service in any country in the European Union (EU) must comply to European regulations. This also holds for medical devices, for which the European Union (EU) Medical Device Regulation (MDR) is applicable [Citation1]. The MDR succeeds the Medical Device Directive (MDD 93/42/EEC) and was fully implemented on 26 May 2021, after a delay of one year due to the COVID-19 pandemic. Driven by a number of scandals, the MDR is stricter compared to the previous MDD [Citation2–4]. This has consequences for various stakeholders including healthcare professionals that prescribe and manufacture custom-made devices [Citation4,Citation5].
The most concrete effort to support healthcare professionals in dealing with the implementation of the MDR for custom-made medical devices that they manufacture is given by Green et al. [Citation6]. They addressed the consequences for UK-based dental professionals that create custom-made medical devices by means of a stepwise description of ten tasks, explanation of terminology, and reference to MDR sections. However, discussions with occupational therapists working in rehabilitation in The Netherlands (healthcare professionals who are experts in advising and manufacturing custom-made devices) suggested that explanation of the MDR alone is not sufficient to start implementing the MDR in their workflow for prescribing assistive devices, as defined by the Dutch prescription guideline [Citation7,Citation8]. The main challenges occupational therapists reported were the translation of the framework offered by the MDR into concrete guidelines and forms as well as the setup of a quality management system (QMS) that complies with the MDR. On top of that, the occupational therapists also would like to start using rapidly evolving manufacturing techniques such as 3D printing to offer further customization of the medical devices that they prescribe to assist clients in Activities of Daily Living (ADL). However, the introduction of these manufacturing techniques raises additional questions regarding compliance with the MDR. Within this study, the challenges and questions of the occupational therapists in the Netherlands were addressed by:
Organizing a series of workshops to offer occupational therapists insight into the rules, regulations and the mindset enforced by the MDR to ensure safe use of custom-made assistive (medical) devices, and
Generating and evaluating a roadmap consisting of practical guidelines and forms for implementation of the MDR as integral part of their current workflow.
Before the method is described in detail, relevant definitions and explanations regarding the MDR are described in line with the approach Green et al. [Citation6].
Medical device regulation for “custom-made medical devices”
This section introduces important definitions related to the MDR requirements for occupational therapists. Special attention is paid to the concepts of “adaptable medical devices” and “custom-made devices”, which are the predominant types of the medical devices that occupational therapists prescribe and develop in co-creation with their clients. As indicated, the Medical Device Regulation (MDR-2017/745) is intended for medical devices, which is defined in Article 2.1 of the MDR as follows [Citation1]:
Medical device means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease, diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability, investigation, replacement or modification of the anatomy or of a physiological or pathological process or state, providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations, and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.
The following products shall also be deemed to be medical devices:
devices for the control or support of conception; products specifically intended for the cleaning, disinfection or sterilization of devices as referred to in Article 1(4) and of those referred to in the first paragraph of this point.
In addition to the definition of a medical device and the determination that the majority of assistive devices can be categorized in Class I, a clear distinction must be made between adaptable medical devices and custom-made medical devices. To this end, Article 2.3 of the MDR defines [Citation1]:
Custom-made device means any device specifically made in accordance with a written prescription of any person authorized by national law by virtue of that person’s professional qualifications which gives, under that person’s responsibility, specific design characteristics, and is intended for the sole use of a particular patient exclusively to meet their individual conditions and needs.
Examples of custom-made medical devices are dental crowns, hand prostheses with specific functionalities [Citation10] or 3D printed cupholders customized to individual clients’ needs such as shown in . In these cases, the level of personalization is embedded right at the start of the design process. For instance, in case of a dental crown, one needs a CT scan or another medical image of that individual crown in order to manufacture this specific crown. In other words, this medical device cannot be made via mass production. The 3D printed cupholder mentioned above was designed by an occupational therapist in co-creation with one particular client. In accordance, this cupholder was manufactured with a written description. In accordance with the MDR, healthcare professionals that design and manufacture custom-made medical devices are considered the legal manufacturer and therefore are legally bound to comply with the MDR. Although fewer MDR rules apply for custom-made medical devices, the implementation of them does have implications for the conventional way of working for occupational therapists. In particular, manufacturers of custom-made medical devices must comply to the following requirements [Citation10] (and double checked by the senior quality manager with the MDR (IdJ) [Citation1]):
Figure 1. Custom-made 3D printed cupholder which has one ear and one stick and is held by the client. Photographer: Inge Hondebrink

Manufacturers shall establish, document, implement and maintain a QMS. No certification of this QMS is required except for manufacturers of class III devices.
Manufacturers must appoint a person responsible for regulatory compliance (PRRC).
Manufacturers must show that the devices are in compliance with the General Safety and Performance Requirements (GSPR) as listed in Annex I of the MDR and sufficient evidence should be provided to demonstrate this ( and Supplementary material Template (MDR classification & conformity)).
Manufactures must show evidence of production, manufacture, design and performance of the device according to design specification (see Supplementary material Template (Production form)). This documentation shall be kept for a period of at least 10 years.
All devices which are put into use must follow the assessment route for custom-made devices as described in Annex XIII of the MDR [Citation1]. This includes a statement containing information about the manufacturer, the person who issued the prescription and the patient of the particular custom-made medical device ( and Supplementary material Template (Declaration of conformity) & Template (Delivery document)).
Manufacturers shall implement a post-market surveillance system and establish appropriate communication channels with relevant healthcare providers/professionals or patients to receive feedback on the quality, (clinical) performance and safety of the devices in the field (see Supplementary material Template (Evaluation form)).
Serious incidents and corrective actions must be reported to the competent authorities.
Figure 2. Screenshot of an example form to document the intended use and argue the classification of the medical device according to Annex VII of the MDR [Citation1].
![Figure 2. Screenshot of an example form to document the intended use and argue the classification of the medical device according to Annex VII of the MDR [Citation1].](/cms/asset/0fe6cb58-ed28-404d-8403-9fc16056a01f/iidt_a_2187889_f0002_c.jpg)
Figure 3. Screenshot of an example form to document compliance of requirements with the MDR, and reference to several ISO standards where applicable to show this compliance.
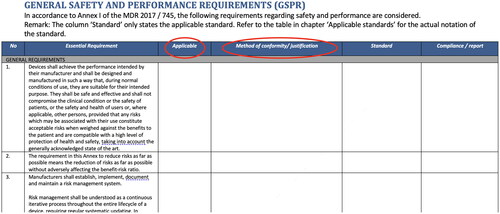
Figure 4. Screenshot of an example form to document the risks and mitigating measure taking, as well as the declaration that with the remaining risks it is safe to use the medical device for its intended use taking into account to set instructions for use.
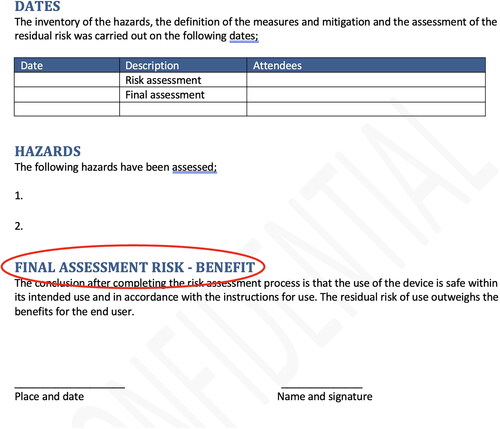
Figure 5. Screenshot of an example form to state that the design and manufacturing of the custom-made device has been executed according to the MDR.

The requirements that do not have to be met are [Citation10]:
Requirements regarding a Unique Device Identification for medical device (UDI) registration, assignment and labelling.
The appointed person responsible for regulatory compliance is not required to be registered in European Databank on Medical Devices (EUDAMED) [Citation12].
Approach towards interpretation and implementation MDR
An iterative codesign approach was carried out to generate a set of guidelines and forms that provide healthcare professionals with practical, hands-on tools for adhering to the MDR for custom-made medical devices. First, the focus was on the explanation and interpretation of the framework of the MDR by the senior quality manager to occupational therapists and to collaboratively interpret what this means for custom-made medical devices. Second, forms and guidelines were generated by the participants tailored to the custom-made Class I medical devices. This codesign process involved a group of seven occupational therapists working in three different rehabilitation centres in The Netherlands to bring cases from their own setting in which a custom-made medical device was created. As a central focus for the codesign workshops, one of the cases was selected using the “Personalized medical devices decision tree” available in a report from the Australian Health Department on 3D-printed medical devices [Citation13]. The selected case involved the customization of a cupholder for a child experiencing limitations in the functioning of its left hand due to cerebral palsy (). Details are presented in the Results section.
Based on this case, four online workshops were prepared and moderated by a senior quality manager (IdJ) who is experienced in working according to the ISO 13485 (Quality management system for medical devices, QMS) [Citation14]. A QMS is a set of processes and methods to ensure that the operation and control of the activities performed in the organization are effective, can be monitored and adjusted and recorded in line with ISO 13485. ISO 13485 offers organizations guidelines for setting up a QMS to demonstrate the ability to provide medical devices and related services that consistently meet customer and applicable regulatory requirements. The senior quality manager had been involved in setting up the MDR documentation for various in-house developed medical devices categorized in Classes I until III.
The four iterative workshops, which are detailed below, were designed to both explain and interpret the MDR and its implications, and to collaboratively develop the aforementioned guidelines and forms. The workshops offered ample room for questions, interaction, discussion, homework and on-the-spot adaptations to make sure that the occupational therapists remained engaged, understood the interpretation and were confident that the implementation steps would fit into their workflows. At the end of each workshop, we evaluated the session with the participants, who were asked to share their experiences and their ideas for further improvement of the guidelines and forms. Based on analyses of the video recordings of the workshops and the field notes taken by the researchers, the quality manager adapted the guidelines and forms to create new iterations that were used as input for the next workshop.
In each session, representatives from the three different local rehabilitation centres participated. In addition to the occupational therapists (n = 7), other participants joined the workshops to make sure that a wide range of expertise and stakeholders was present. These included an adaptation technician (n = 1), a medical engineer (n = 1), policy officers (n = 2), (quality) managers (n = 2), a 3D printing expert (n = 1), a lecturer in engineering (n = 2), a student engineer (n = 1), and researchers (n = 4). The organizations, and thus the participants in the workshops, were all partners in an ongoing action research project focusing on 3D printing assistive devices. In each of these organizations, a 3D printer was installed and learning communities were formed to gain hands-on experience with manufacturing 3D-printed assistive devices. In practice, all participants from these organizations were acquainted with the main principles of 3D printing, but still had rather limited experience with designing and printing devices.
Workshop 1 introduction MDR
The first workshop covered the following topics: the definition of a medical device and of custom-made medical devices, classification of medical devices, intended use of a medical device, generic safety and performance requirements, risk analysis and acting upon it, requirements regarding manufacturing, traceability, packaging, labelling, certification of raw materials, the verification of requirements, QMS, quality procedures, registration, declaration of conformity and notification to appointed organizations. This was an informative workshop with mainly Q&A as interactive component.
Workshop 2 what is already documented in the cupholder case
The second workshop covered the inventory of current work flows of the occupational therapists and their organizations regarding documentation and what is relevant for the MDR. More specifically, this included current workflows, documentation of devices handed to clients, identification of persons responsible for quality management, purchasing of raw material, description of requirements, and post-market surveillance testing.
With this inventory, the occupational therapists were asked to do some homework in between the workshops 2 and 3 by filling out a first version of forms prepared by the senior quality manager regarding the case of the cupholder. These forms covered design requirements, production, (mechanical) testing, user testing and a conformity declaration.
Workshop 3 intended use and risk analysis for the cupholder case
Workshop 3 covered a continuation of the cupholder case (). The senior quality manager give advice regarding the documents that were already in place, also he presented modified versions of the forms which were filled out in a proper manner for references. The participants were asked to review them. Subsequently, the focus was on defining the intended use (and possible misuse), performing a risk analysis, acting upon it, and final verification of acceptable risks involving the medical device. In co-creation during the workshop, the intended use was defined; and a risk analysis of the cupholder as prepared by the quality manager was discussed to show what is important to document and to verify the contents for the cupholder case. Based on the input of the workshop, final versions were prepared of all forms for Workshop 4.
Workshop 4 wrap-up and tuning
Based on the results of Workshops 2 and 3, the quality manager adapted the proposed forms with the filled-out documentation of the cupholder case. In a final round, these documents were reviewed by the workshop participants and tuned to fit, resulting in final forms for various steps and a filled-out example of the cupholder case for future reference. In this final workshop, explanation and instructions were given on the documentation of the working process, the mandatory registration with national legal organizations, labelling, post-market surveillance and declaration of conformity, as well as measures participants should arrange within their own organizations, such as appointing a quality officer/responsible person and a digital and safe accessible location to store the documentation.
Results
As indicated the selected case was the customization of a cupholder for a child with cerebral palsy experiencing limitations in the functioning of his left hand (). A conventional ergonomic cupholder with two ears did not meet the client’s hand functioning requirements, since the child was unable to wrap its hand around the left ear of the cupholder. Therefore, a custom-made cupholder was designed with one ear and one handle. Shapes that were specifically modelled according to the dimensions of the client’s hand and its tolerable gripping force. To design the most appropriate shape, a model for the grip was created by the occupational therapist together with the child using modelling clay. This clay model was scanned and transferred into a 3D computer-aided design by an engineer. Subsequently, the design was sliced and 3D printed using a fused deposition modelling (FDM) printing technique in collaboration with a local 3D printing company, RepRapUniverse (Kerkrade, The Netherlands, https://reprapuniverse.com/). The cupholder was printed in polylactic acid (PLA), a commonly used biocompatible 3D printing material.
During the evaluations at the end of each codesign workshops, participants indicated that they found the presentation on the MDR and related processes in Workshop 1 very informative. They indicated that some of the requirements for custom-made medical devices were already common practice, since occupational therapists are experts in advising and adapting mass produced, or creating new tailormade assistive medical devices for their clients’ needs. More specifically, the occupational therapists indicated that it is common practice to formulate design requirements and to evaluate the device performance during check-up visits of the clients. The occupational therapists unfamiliar with the MDR also indicated that the information presented was experienced as complicated and of high density at times. They expressed concerns about the feasibility of complying to Requirements 4 and 6 of the MDR as presented in this paper. The required paperwork to meet the MDR, generated a feeling of “burden” for the occupational therapists. This was driven by the fact that the time that occupational therapists are allowed to spend with each client is limited. Using this feedback, the remaining workshops were modified. More time was spent during Workshops 2 and 3 on explaining that the MDR considers the occupational therapists as manufacturers and a such they are legally bound to the MDR. To facilitate compliance with the MDR, forms were generated and adjusted during Workshops 2 and 3. Final versions were filled out for the cupholder case were agreed upon in Workshop 4 for future reference (see and Supplementary material). Supplementary to these forms, instructions were generated, indicating when, who and what should be filled out. Taking into account the occupational therapists’ concerns about feasibility, it was stressed how to proceed as efficiently as possible. Some requirements for instance involve actions that each organization only has to perform once. Specifically, Requirements 1, 2 and 7 from the section Medical Device Regulation for “custom-made medical devices” of this paper. These include:
Table 1. Overview of forms, the reference to the supplementary material (templates) and short description of the content.
Requirement 1: Generate a main document (in an accessible digital environment) and inform co-workers that custom-made medical devices are designed and manufactured according to the procedure as defined by the “Procesbeschrijving Hulpmiddelenzorg” [Citation7]. From that main document point to relevant forms (see and Supplementary material) that need to be completed to show that indeed the development and manufacturing are performed according to that procedure.
Requirement 2: Appoint a responsible (quality) manager who oversees and coordinates the documentation regarding cusom-made medical devices. The (quality) manager assures completeness of the documentation for a custom-made medical device and verifies archiving of the documentation for at least ten years (Annex XIII of the MDR) [Citation1].
Requirement 7: Sign up as manufacturer at legally assigned organization in the country of practice (in the Netherlands at https://www.farmatec.nl/medische-hulpmiddelen – “eHerkenning inlogmiddel aanvragen”).
Furthermore, several forms (specifically those meeting Requirements 3–6 in this paper) can be reused or modified with minor changes for similar custom-made medical devices. As such, standard documentation can be created and re-used for medical device classes that have similar intended uses; covered by a product category. For example, if another client needs yet another custom-made design of the cupholder as shown in , for example with two side bars, the “intended use” is the same. For such a product category, the Forms 1, 2, 3 and 5 from can be re-used.
The Declaration of conformity serves two purposes. The first is to notify this product category of medical devices with the competent national authorities of the country of practice. In the Netherlands, this authority is Inspectie Gezondheidszorg en Jeugd (IGJ, www.igj.nl). (https://www.farmatec.nl/medische-hulpmiddelen/medische-hulpmiddelen-naar-maat). The second is when the device is handed to the client with details of the specific patient are being filled out.
Finally, Forms 4–7 () need to be filled out for each individual custom-made medical device (Annex XIII of MDR [Citation1]).
(https://www.igj.nl/zorgsectoren/medische-technologie/toezicht-op-producten/vigilantie-medische-technologie/melden-als-fabrikant). Note that Form 7 (Template (Evaluation)) is set up as a post-market surveillance tool for each individual custom-made device. This way the occupational therapists in their role of legal manufacturer can monitor the performance of the device.
From our analyses of the assignments and the participants’ input, several mismatches and misunderstandings were noticed between the terminology used by the occupational therapists and the quality manager and engineers. As a result, some forms were not completed, because the participants did not understand what to fill out. In some instance, even when explained, occupational therapists did not fully understand or did not feel responsible for filling out a form, because they lacked sufficient training. Participants indicated several times during the workshops that when using 3D printing technology to manufacture a custom-made device, collaboration with engineers, expert designers and/or 3D manufacturers is essential. These may be experts within their own organization, such as an adaptation technician but may also be experts from an external company. In the latter case, such companies act as an outsourcing partner. They could be asked to provide the required qualifications (e.g. certification of technology, production procedures and use of certified materials) to make sure that the products meet the technical specifications. In view of the MDR, the occupational therapist remains the legal manufacturer, so it is their responsibility to request proof that the work done by the outsourcing party is performed according to the MDR. Such companies are usually more capable in engineering, because they have designers employed whose specific task it is to design and manufacture products. If certified for ISO 13485 or ISO 9001, they already have QMS in place. So, this could be a fruitful and safe way to implement the MDR in the field of occupational therapy. The CEO of the participating 3D printing company, who participated in the workshops, mentioned to have recently started a certification procedure for his equipment and standardized working procedures in order to become a legal medical device manufacturer.
Discussion
The results of the workshops show that the MDR is complex to understand and that guidance is needed by experts familiar with the MDR for healthcare professionals; such as occupational therapists working in rehabilitation settings when manufacturing custom-made medical devices. At the start of the workshops, participants, including engineers and managers, indicated limited knowledge of the meaning and the impact of the MDR for their organizations and work. This illustrates the need for support to create MDR-compliant assistive devices in rehabilitation settings.
One of the conclusions of all participants was that they confirmed the added value in using 3D printing technology to manufacture customized devices for clients in rehabilitation, because products available on the market often do not fit the individual needs and wishes of clients. However, all participants stated that close collaboration between occupational therapists, their clients and engineers, designers and/or 3D printing experts is required during the process of manufacturing customized 3D printed assistive devices. Occupational therapists are experts in development of creative assistive solutions for functional problems their clients experience in daily life, but they lack essential technical knowledge and skills that are needed for designing and manufacturing 3D-printed objects. Designers and engineers on the other hand lack clinical expertise, and are not necessarily familiar with MDR requirements, especially when they do not specialize in manufacturing medical devices. As such, even though collaborations between clinicians, designers and engineers may yield the required technical and clinical skills to manufacturing 3D-printed assistive devices, still support is required on the quality management side to comply with requirements of the MDR. This includes risk analyses and quality testing of 3D designs and production. The forms developed during the course of the four workshops presented in this paper are a first step in offering such support. For larger care organizations offering custom-made assistive devices, it is strongly recommended to appoint a dedicated (quality) manager, who can lead and monitor the entire process and makes sure that documentation is done in accordance with the MDR.
Regarding the process and the forms that were developed during the workshops, the participants with a background in occupational therapy mentioned that the proposed way of working is very extensive and time consuming in proportion to the number of requests for assistive devices for which 3D printing might be a suitable option. Occupational therapists seemed especially reluctant towards the extensive documentation that they should record for each and every individual assistive device they manufacture. Therefore, the authors decided to develop a practical guideline and accompanying forms that can accommodate product categories of assistive devices instead of a single one. Such product categories consist of devices with similar characteristics and manufacturing techniques, for which the authors developed dedicated forms. For instance, one category of assistive devices consists of geometric shapes to form “customizable grips” for personalization of cutlery, writing aids, and household appliances. Following the guideline of meeting the Requirements 1–7 in this paper, forms were generated to fulfil these requirements with a clear indication that for such a product category of assistive devices certain forms need be filled out only once for the entire category, and other forms need to be fill out for each individual device that is given to the client. This should – in time – significantly reduce the documentation workload for newly printed assistive devices.
Despite the measures the authors took to reduce the workload, compliance with the MDR while manufacturing 3D-printed assistive devices (and thus qualify as custom-made medical device) still implies time and effort on behalf of the legal manufacturer. Although the MDR aims to ensure and improve the quality and safety of medical devices and to prevent harmful consequences, the requirements (and the accompanying extensive documentation/procedure) of the MDR were perceived as excessive for certain kinds of assistive devices, especially for low-risk Class I medical devices, such as the customizable grips mentioned above. As such, some of the occupational therapists who participated in the workshops were rather demotivated by learning about the MDR requirements, which in some cases even made them doubt as to whether they should continue using 3D printing technology altogether. However, occupational therapists also realized that this is not really a solution, as the MDR does not only apply to 3D-printed assistive devices, but to any custom-made medical device prescribed by an occupational therapist. Occupational therapists traditionally are the experts on advising and supplying assistive devices to support client’s independent living and participation in daily activities. In case of no sufficient device being available on the market, occupational therapists are the ones to create custom-made medical devices using many different methods and materials (such as wood or foam rubber). Occupational therapists, nationally and across Europe, have been creating custom-made medical devices for decades. The MDR forces them together with their organizations to adapt their working procedures in order to guarantee clients’ safety. Form 7 () is a questionnaire commonly used to evaluate mass-produced adaptable medical devices. In this paper, this form is suggested to be used as a post-market surveillance tool for each individual custom-made device. So, the focus is on the performance and safety of the product and as such this form is not part of a clinical investigation. Therefore, it is important to continue the search for ways to make the process of MDR-compliant manufacturing of assistive devices accessible and achievable for collaborating clinicians and designers and/or engineers. Besides for rehabilitation centres, such a procedure is also relevant for self-employed occupational therapists, or other clinicians who might want to be involved in creating customized devices for their clients.
Besides making the documentation process easier and faster (see Supplemental material), as was aimed for by the forms, another way of lowering the threshold to developing custom-made medical assistive devices could be to share 3D designs for re-use. Open source databases such as Thingiverse.com already offer a significant amount of 3D models for assistive devices that occupational therapists could print or adapt for their own clients. An interesting way forward regarding MDR-compliant custom-made assistive devices in this respect could be shared MDR documentation in addition to the digital design files.
Conclusion
Custom-made medical devices in Europe must be produced and provided in accordance with the Medical Device Regulation. This paper offers interpretation and practical guidelines to implement the requirements for custom-made devices as prescribed and manufactured by occupational therapists in cocreation with clients and in close collaboration with engineers, designers and 3D printing experts.
Based on the experiences of the occupational therapists who participated in our workshops, it is highly recommended to work in collaboration with designers and engineers who are experts in designing and manufacturing, and who can assist in prototyping, testing and documentation, or who are fully specialized in producing this type of medical devices.
Template__Declaration_of_conformity_.docx
Download MS Word (50.8 KB)Template__Risk_Assessment_File_.docx
Download MS Word (55.9 KB)Template__Risk_Analysis_File_.xlsx
Download MS Excel (69.6 KB)Template__Requirements_.docx
Download MS Word (55.6 KB)Template__MDR_classification___conformity_.docx
Download MS Word (108.2 KB)Acknowledgements
We would like to thank all the participants of Sevagram, Adelante Rehabilitation Centre, Libra Rehabilitation & Audiology, Zuyd University of Applied Sciences and RepRapUniverse for their contribution and valuable input during the four workshops.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, 2017; [cited 2022 Mar 23]. Available from: https://eur-lex-europa-eu.ezproxy2.utwente.nl/legal-content/EN/TXT/HTML/?uri=CELEX:32017R0745&from=IT#d1e1058-1-1.
- Martindale V, Menache A. The PIP scandal: an analysis of the process of quality control that failed to safeguard women from the health risks. J R Soc Med. 2013;106(5):173–177.
- Cohen D. Faulty hip implant shows up failings of EU regulation. Br Med J. 2012;345:e7163.
- Vasiljeva K, van Duren BH, Pandit H. Changing device regulations in the European union: impact on research, innovation and clinical practice. Indian J Orthop. 2020;54(2):123–129.
- Green JI. The impact of medical device regulation on hospital doctors who prescribe and manufacture custom-made devices. Br J Hosp Med. 2020;81(12):1–6.
- Green JIJ. Medical device regulation: requirements for dental professionals who prescribe and manufacture custom-made devices. Prim Dent J. 2021;10(1):64–88.
- CVZ. Procesbeschrijving Hulpmiddelenzorg. 2010.
- Heerkens Y, Bougie T, Claus E. The use of the ICF in the process of supplying assistive products: discussion paper based on the experience using a general dutch prescription guideline. Prosthet Orthot Int. 2011;35(3):310–317.
- CE Tool: Panton, Holland Innovative, Fris & Fruitig. 2022; [cited 2022 Apr 22]. Available from: https://cetool.nl/en.
- Questions and Answers on Custom-Made Devices. Medical Device Coordination Group. 2021; [cited 2022 Apr 22]. Available from: https://ec.europa.eu/health/system/files/2021-03/mdcg_2021-3_en_0.pdf.
- MDR requirements for custom-made medical devices: MDR Regulator; 2022; [cited 2022 Apr 22]. Available from: https://mdrregulator.com/news/custom-made-medical-devices-mdr.
- EUDAMED – European Database on Medical Devices. European Commission. 2021; [cited 2022 Mar 23]. Available from: ec.europa.eu/tools/eudamed.
- Personalised medical devices (including 3D-printed devices). 5 ed. Australia: Therapeutic Goods Administration. 2021. p. 32; [cited 2022 Apr 22]. Available from: https://www.tga.gov.au/sites/default/files/personalised-medical-devices-including-3d-printed-devices.pdf.
- ISO 13485 – Medical Devices – Quality management systems – requirements for regulatory purposes, 3 ed. International Organization for Standardization, Geneva, Switzerland, 2016; [cited 2022 Apr 22]. Available from: https://www.iso.org.