
ABSTRACT
Introduction: Hereditary transthyretin-mediated amyloidosis (ATTRv; v for variant) is an underdiagnosed, progressive, and fatal multisystemic disease with a heterogenous clinical phenotype that is caused by TTR gene mutations that destabilize the TTR protein, resulting in its misfolding, aggregation, and deposition in tissues throughout the body.
Areas covered: Inotersen, an antisense oligonucleotide inhibitor, was recently approved in the United States and Europe for the treatment of the polyneuropathy of ATTRv based on the positive results obtained in the pivotal phase 3 trial, NEURO-TTR. This review will discuss the mechanism of action of inotersen and its pharmacology, clinical efficacy, and safety and tolerability. A PubMed search using the terms ‘inotersen,’ ‘AG10,’ ‘antisense oligonucleotide,’ ‘hereditary transthyretin amyloidosis,’ ‘familial amyloid polyneuropathy,’ and ‘familial amyloid cardiomyopathy’ was performed, and the results were screened for the most relevant English language publications. The bibliographies of all retrieved articles were manually searched to identify additional studies of relevance.
Expert opinion: Inotersen targets the disease-forming protein, TTR, and has been shown to improve quality of life and neuropathy progression in patients with stage 1 or 2 ATTRv with polyneuropathy. Inotersen is well tolerated, with a manageable safety profile through regular monitoring for the development of glomerulonephritis or thrombocytopenia.
1. Introduction
Transthyretin amyloidosis (ATTR) is an underdiagnosed, progressive, fatal multisystemic disease that results from the misfolding, aggregation, and deposition of the plasma transport protein, transthyretin (TTR), in various tissues through the body [Citation1–Citation4]. ATTR may be acquired, known as wild-type ATTR, or hereditary (ATTRv; v for variant, formerly abbreviated hATTR), which develops as a result of mutations in the TTR gene that destabilize the resultant TTR protein [Citation1].
ATTRv often presents as a variable clinical phenotype because of amyloid deposition in multiple organ systems () [Citation2,Citation5–Citation7]. Deposition is most common in the peripheral nerves and heart, resulting in polyneuropathy and cardiomyopathy, respectively [Citation8–Citation10]. If untreated, symptoms of ATTRv are generally progressive until death, which typically occurs 3–15 years after presentation [Citation5]. The current estimated global prevalence of ATTRv with polyneuropathy or cardiomyopathy is approximately 10,000 and 40,000 persons, respectively [Citation8,Citation11].
Figure 1. Clinical manifestations associated with hereditary transthyretin amyloidosis (ATTRv) [Citation2,Citation5–Citation7]. ATTRv is a progressive, multisystemic disease characterized by the deposition of amyloid fibrils in various organs and tissues throughout the body, including the nerves, heart, gastrointestinal tract, liver, and kidney. CNS, central nervous system.
Adapted with permission from Conceição I, González-Duarte A, Obici L, et al. ‘Red-flag’ symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9.
![Figure 1. Clinical manifestations associated with hereditary transthyretin amyloidosis (ATTRv) [Citation2,Citation5–Citation7]. ATTRv is a progressive, multisystemic disease characterized by the deposition of amyloid fibrils in various organs and tissues throughout the body, including the nerves, heart, gastrointestinal tract, liver, and kidney. CNS, central nervous system.Adapted with permission from Conceição I, González-Duarte A, Obici L, et al. ‘Red-flag’ symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9.](/cms/asset/dba10662-c830-4c38-9b1c-b07c16d61f43/ierj_a_1635008_f0001_b.gif)
TTR is a 55-kDa transporter protein primarily synthesized and secreted by the liver [Citation1,Citation2,Citation12–Citation14]. The main function of TTR in plasma is to transport the thyroid hormone, thyroxine, and the retinol (vitamin A)/retinol-binding protein (RBP) complex [Citation1,Citation2,Citation12]. In its native state, TTR exists as a soluble homotetrameric complex comprising four single-chain TTR monomers [Citation1,Citation2]. When the TTR gene is mutated, the tetrameric complex is destabilized and dissociates into monomers, resulting in the formation and deposition of amyloid fibrils and the disease known as amyloidosis [Citation2]. More than 120 genetic variants of the TTR gene exist that result in ATTRv, with some mutations being more strongly associated with polyneuropathy (e.g. Val30Met) and others with cardiomyopathy (e.g. Val122Ile) [Citation1,Citation2,Citation13,Citation14]. Val30Met is the most common variant worldwide and is found with higher prevalence in Portugal, Spain (Majorca), Brazil, France, Japan, and Sweden, while Val122Ile is most commonly found in African Americans [Citation1,Citation14].
Recently, two new first-in-class drugs, inotersen (Tegsedi, Akcea Therapeutics, Inc.) and patisiran (Onpattro, Alnylam Pharmaceuticals, Inc.), have been approved in the United States for the treatment of polyneuropathy of ATTRv in adult patients and in Europe for the treatment of stage 1 or 2 polyneuropathy in adults with ATTRv [Citation15–Citation18]. These novel therapies reduce the plasma concentration of both variant and wild-type TTR by targeting its mRNA. This article will review inotersen, an antisense oligonucleotide (ASO) inhibitor, for the treatment of polyneuropathy caused by ATTRv, focusing on the mechanism of action of inotersen and its pharmacology, clinical efficacy, and safety and tolerability.
2. Overview of the market
Until recently, there were no US Food and Drug Administration (FDA)-approved treatment options available for ATTRv. In patients with early-stage ATTRv, orthotopic liver transplantation (OLT) has been shown to slow the progression of neuropathy and improve survival; however, use of OLT is limited by organ availability, cost, the need for lifelong immunosuppression, and surgical morbidity and mortality [Citation5,Citation19–Citation21]. Heart transplantation with or without OLT may also be considered for ATTRv with significant cardiac involvement but has similar limitations as OLT [Citation1,Citation5,Citation22].
An alternative strategy for the treatment of ATTRv is to stabilize the TTR protein tetramer, preventing the dissociation of misfolded TTR into monomers and thus the formation of amyloid fibrils [Citation5]. Diflunisal, a nonsteroidal anti-inflammatory drug, prevents the formation of TTR amyloid fibrils [Citation1]. A randomized, double-blind, placebo-controlled phase 3 study demonstrated that twice-daily diflunisal 250 mg over 2 years significantly reduced the rate of progression of neurologic impairment and preserved quality of life (QoL) in patients with ATTRv [Citation23]. Although diflunisal is not FDA approved for the treatment of ATTRv, it is available as an off-label option in the United States and in some European countries [Citation5]. However, long-term use of nonsteroidal anti-inflammatory drugs is associated with an increased risk of gastrointestinal, renal, and cardiac toxicity; as a result, caution should be taken when considering the use of diflunisal in ATTRv patients with cardiac involvement [Citation8].
Tafamidis is an oral, small-molecule TTR stabilizer that is approved in Europe and selected countries in Asia and Latin America for the treatment of ATTRv in adults with stage 1 symptomatic polyneuropathy to delay peripheral impairment [Citation24–Citation26]; tafamidis was not approved for neuropathy by the FDA because of inadequate evidence of efficacy [Citation8,Citation26]. In the randomized, placebo-controlled phase 2/3 Fx-005 study, tafamidis therapy demonstrated TTR stabilization in 98% of patients, reduced neurologic deterioration, preserved nerve function, and maintained QoL in patients with early-stage Val30Met ATTRv [Citation27]. Additional analyses and long-term follow-up of this pivotal trial further supported the efficacy and safety of tafamidis in patients with early-stage ATTRv-associated polyneuropathy [Citation28,Citation29], and a recent analysis of the Transthyretin Amyloidosis Outcomes Survey (THAOS) reported the ability of tafamidis to delay disease progression during 2 years of follow-up [Citation30]. Recently, the phase 3 ATTR-ACT trial reported that tafamidis was associated with reductions in all-cause mortality and cardiovascular-related hospitalizations, along with a reduced decline in functional capacity and QoL in patients with cardiomyopathy associated with either ATTRv or wild-type ATTR [Citation31]. As a result of these findings, tafamidis was granted fast-track designation by the FDA for cardiomyopathy-associated ATTR. Tafamidis meglumine (Vyndaqel, Pfizer Inc.) and tafamidis (Vyndamax, Pfizer Inc.) were recently approved for the treatment of cardiomyopathy of wild-type ATTR or ATTRv in adults to reduce cardiovascular mortality and cardiovascular-related hospitalization [Citation32].
The TTR-lowering agents inotersen and patisiran are the first agents to target TTR, the disease-forming protein of ATTRv. Patisiran is a double-stranded small interfering RNA administered via intravenous infusion that targets a sequence on TTR messenger RNA to decrease production of TTR. It was recently approved by the FDA for the treatment of polyneuropathy of ATTRv in adult patients [Citation15], and in Europe for the treatment of ATTRv in adults with stage 1 or 2 polyneuropathy [Citation16] based on the findings of the large randomized, double-blind, placebo-controlled phase 3 APOLLO trial that showed a therapeutic benefit in polyneuropathy, neuropathy, and QoL with patisiran versus placebo in patients with ATTRv [Citation33]. The ASO, inotersen, is the focus of this review and will be discussed in detail below.
Other agents currently undergoing clinical development for the treatment of ATTRv include novel TTR stabilizers that offer greater stability of TTR tetramers (e.g. tolcapone [SOM0226], CSP-1103 [CHF5074], AG10) [Citation34–Citation40], combination therapy with doxycycline (disrupts TTR amyloid fibril formation) and tauroursodeoxycholic acid (reduces nonfibrillar TTR deposition) [Citation41,Citation42], and anti-TTR monoclonal antibodies that target amyloid monomers and inhibit fibril formation and promote phagocytosis of TTR aggregates [Citation43–Citation45]. While these agents show promise, published data from randomized controlled trials are currently lacking.
3. Introduction to inotersen
3.1. Chemical property
Inotersen sodium, a 2ʹ-O-methoxyethyl-modified second-generation ASO, is formulated as a white to pale-yellow solid that is freely soluble in water and in phosphate buffer (pH 7.5 to 8.5) [Citation46,Citation47]. The molecular formula of inotersen sodium is C230H299N69Na19O121P19S19, and its molecular weight is 7600.73 Da [Citation47].
3.2. Mechanism of action
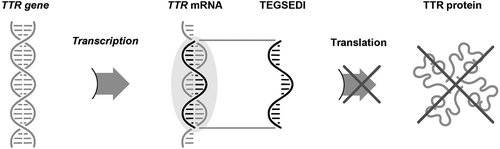
Inotersen was designed to inhibit the hepatic production of mutant and wild-type TTR () [Citation46]. Using a Watson-Crick hybridization binding mechanism, inotersen targets TTR mRNA produced in the nucleus of hepatocytes, resulting in the degradation of TTR RNA, which in turn prevents the translation of TTR mRNA into TTR protein [Citation46]. The binding site on inotersen occurs in a 3′-untranslated region of the TTR mRNA that is devoid of known TTR mutations; thus, its action prevents the production of both mutant and wild-type TTR by the liver [Citation46].
Figure 2. Mechanism of action of inotersen. mRNA, messenger ribonucleic acid; TTR, transthyretin.
Reprinted with permission from Benson MD et al. Neurodegener Dis Manag. 2019;9:25–30.
3.3. Pharmacodynamics
In vitro studies in hepatocyte-like cell lines (HLCs) derived from pluripotent stem cells from patients with ATTRv showed that the administration of inotersen downregulated TTR mRNA in HLCs, as well as monomeric and dimeric forms of TTR protein in HLC cell culture supernatant by 80–90% [Citation48]. Inotersen has also been evaluated in vivo in transgenic mice and cynomolgus monkeys. In transgenic mice harboring the TTR Ile84Ser mutation, subcutaneous (SC) injection of inotersen at doses ranging from 10 to 100 mg/kg/week for 4 weeks was associated with >90% reduction in hepatic TTR mRNA and plasma TTR protein levels at the highest doses tested, compared with phosphate buffered saline-treated controls; these reductions were directly proportional to inotersen accumulation in the liver [Citation49]. SC administration of inotersen 25 mg/kg (3 times during week 1, then twice weekly during weeks 2–12) in cynomolgus monkeys was well tolerated and was associated with a reduction in hepatic TTR mRNA levels of approximately 90% and a mean reduction in plasma TTR protein levels of approximately 80% at 12 weeks of treatment [Citation49]. Taken together, these preclinical studies highlighted the ability of inotersen to provide substantial reductions in both wild-type and mutant TTR levels.
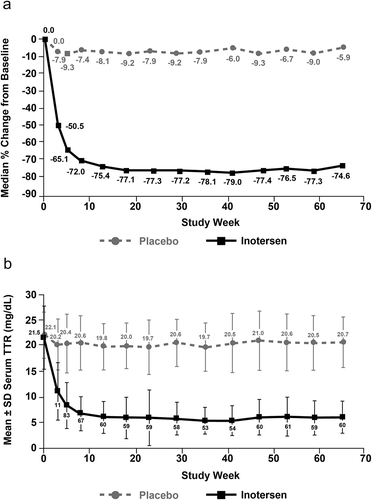
Consistent with preclinical findings, inotersen was also associated with reductions in both wild-type and mutant forms of TTR in humans. In healthy human subjects, dose-dependent reductions in plasma TTR protein levels were observed in a phase 1 trial, with mean percentage reductions from baseline of 77% and 79% for the 300- and 400-mg dose levels after 4 weeks of treatment [Citation49]. In the 15-month, phase 3 NEURO-TTR pivotal trial (NCT01737398; ), once-weekly SC administration of inotersen 300 mg (equivalent to 284 mg free acid) achieved steady-state concentrations of inotersen in the liver at approximately 13 weeks, which resulted in a median percentage reduction from baseline in serum TTR levels of 79.0% during weeks 13–65 of treatment () [Citation50].
Figure 3. Study design of NEURO-TTR and the open-label extension.
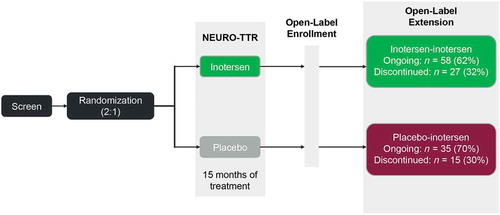
Figure 4. Serum transthyretin (TTR) over time. (a) Median percentage change from baseline and (b) mean absolute TTR level.
Reprinted with permission from Benson MD et al. N Engl J Med. 2018;379:22–31.
Another pharmacodynamic effect observed following inotersen therapy is the change in plasma levels of vitamin A. Because serum TTR is a carrier of retinol binding protein 4 (RBP4), the main carrier of retinol (vitamin A) in the blood, inotersen reduces plasma vitamin A levels to below the lower limit of normal [Citation47,Citation49,Citation51].
3.4. Pharmacokinetics and metabolism
Subcutaneous administration of inotersen is associated with dose-dependent pharmacokinetics (PK) [Citation47,Citation51]. Following administration, inotersen is rapidly absorbed into the systemic circulation, and peak plasma concentrations are achieved after a median of 2–4 h [Citation47,Citation49,Citation51]. More than 94% of inotersen is bound to plasma protein, regardless of drug concentration, and extensive distribution into tissues at steady state is observed in patients with ATTRv amyloidosis, with a volume of distribution of 293 l [Citation47,Citation51].
Inotersen is metabolized by endonucleases in tissues, which leads to the formation of shorter inactive oligonucleotides that are then metabolized by exonucleases [Citation47,Citation51]. Inotersen is not a substrate for cytochrome (CYP) P450 oxidation. Following SC administration, the unchanged drug is the main circulating component, with <1% of unchanged drug being excreted in the first 24 h. Inotersen and its oligonucleotide metabolites are excreted in the urine. The elimination half-life of inotersen following SC administration is approximately 1 month.
Population PK analyses have shown that age, body weight, sex, and race have no clinically relevant effects on inotersen exposure [Citation47,Citation51]. There are currently no data on inotersen in patients with severe renal impairment, end-stage renal disease, moderate or severe hepatic impairment, or prior liver transplantation.
3.5. Drug interactions
No studies have been conducted to assess the potential clinical drug interactions with inotersen [Citation47,Citation51]. Inotersen is not a substrate for inhibitors/inducers of CYP enzymes or major transporters [Citation47]. In a population PK analysis, the concomitant use of diuretics, antithrombotics, and analgesics did not impact the PK parameters of inotersen [Citation47].
4. Clinical efficacy
Inotersen has been evaluated in a phase 1 trial, an investigator-initiated phase 2 trial, and a pivotal phase 3 trial, which are summarized in .
Table 1. Clinical trials of inotersen.
4.1. Phase 1 study
In a placebo-controlled phase 1 study, 65 healthy volunteers received placebo or inotersen (50–400 mg, SC) as single doses (n = 16) or multiple doses (n = 49) [Citation49]. Subjects in the multiple-dose cohort received a total of six doses; three doses on alternate days during the first week (days 1, 3, and 5), followed by once-weekly doses for 3 weeks (days 8, 15, and 22) [Citation49].
In the multiple-dose cohorts, inotersen was associated with mean percentage reductions in circulating serum TTR levels from baseline to day 29 that ranged from 8% with the 50-mg dose to 76% with the 400-mg dose (all p ≤ 0.001 vs. baseline except for the 50-mg dose [p = 0.07]). The reduced serum TTR levels were maintained for approximately 3–4 weeks after the last dose, gradually returning to baseline values. In the 300- and 400-mg inotersen cohorts, mean serum TTR levels remained 30% below baseline levels at 10 weeks after the last dose. Inotersen also lowered serum RBP4 levels, which were significantly reduced from baseline to day 29 in a dose-dependent manner (from 11% to 75%; all p < 0.05). The changes in TTR levels were significantly correlated with changes in RBP4 levels (R2 = 0.88; p < 0.0001).
4.2. Phase 3 study
The NEURO-TTR trial was an international, randomized, double-blind, placebo-controlled, 15-month pivotal phase 3 trial conducted at 24 centers in 10 countries () [Citation50]. Patients were included who were aged 18–82 years with a diagnosis of stage 1 or 2 ATTRv with polyneuropathy (Neuropathy Impairment Score [NIS] of 10 to 130); all patients had a TTR mutation determined by genotyping and biopsy-proven amyloid deposits. Recipients of liver transplantation were not eligible for the trial. Eligible patients were randomly assigned 2:1 to SC inotersen 300 mg or placebo. To achieve steady-state drug levels, 3 injections were administered during the first week, followed by once-weekly injections for the next 64 weeks. The coprimary endpoints were the change from baseline to week 66 in the standardized modified NIS Score+7 (mNIS+7; range, −22.3 to 346.3, with higher scores indicating more impairment and a 2-point change defined as a minimum clinically meaningful detectable change) composite score and the total score on the Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN; range, −4 to 136, with higher scores indicating worse QoL) questionnaire. Secondary and tertiary efficacy endpoints included components and subcomponents of the mNIS+7 test, the Norfolk QoL-DN questionnaire according to domain, and the 36-item Short-Form Health Survey (SF-36).
Of the 173 patients who were randomized in the NEURO-TTR trial, 172 received at least one dose of study treatment; 112 patients were assigned to the inotersen group and 60 to the placebo group [Citation50]. Overall, 139 out of 172 patients (81%) completed the 15-month treatment period; adverse events (AEs) were the main reason for discontinuation of study treatment in the inotersen group (16/112, 14%). Patients who completed this 15-month trial were eligible to enter the open-label extension (OLE) study.
Baseline characteristics were well-balanced between the two groups [Citation50,Citation52]. In the overall study population, the mean age was 59 years, 69% were male, and 92% were white. Fifty-two percent of patients carried the Val30Met mutation (out of 27 different TTR mutations), 67% had stage 1 disease, 58% had previously received tafamidis or diflunisal, and 63% had cardiomyopathy (defined as a diagnosis of TTR cardiomyopathy at study entry or the presence of an interventricular wall thickness of ≥13 mm on thoracic echocardiography at baseline and no known history of persistent hypertension within 12 months of screening). In a recent evaluation of the disease characteristics of ATTRv patients in the NEURO-TTR trial, the mean duration of neuropathy symptoms was 5.3 years, and the symptoms were shown to affect multiple organs and systems, with nearly 70% of patients showing widespread involvement with weakness, sensory loss, and autonomic disturbance [Citation52]. The baseline data from NEURO-TTR highlight that ATTRv is a multisystemic disease, with most patients presenting with both polyneuropathy and cardiomyopathy.
Once-weekly treatment with SC inotersen 300 mg for 15 months in the NEURO-TTR trial significantly improved neuropathy symptoms and health-related QoL, compared with placebo [Citation50]. At baseline, the mean (± standard deviation [SD]) mNIS+7 composite score was 79.2 ± 37.0 in the inotersen group and 74.8 ± 39.0 in the placebo group, and mean Norfolk QoL-DN total scores were 48.2 ± 27.5 and 48.7 ± 26.7, respectively. The difference in the least-squares mean (LSM) change from baseline to week 66 in the mNIS+7 composite score between the inotersen and placebo groups was −19.7 points (95% confidence interval [CI], −26.4 to −13.0; p < 0.001) in favor of inotersen (). Similarly, the difference in the LSM change from baseline to week 66 in Norfolk QoL-DN total scores between the inotersen and placebo groups was −11.7 (95% CI, −18.3 to −5.1; p < 0.001) in favor of inotersen (). These significant improvements in the coprimary endpoints of NEURO-TTR were independent of disease stage, mutation type, or the presence of cardiomyopathy.
Figure 5. Coprimary endpoints from the pivotal phase 3 NEURO-TTR trial [Citation47,Citation50]. (a) Change from baseline to week 66 in the mNIS+7 composite score, and (b) Change from baseline to week 66 in the Norfolk QoL-DN total score.
![Figure 5. Coprimary endpoints from the pivotal phase 3 NEURO-TTR trial [Citation47,Citation50]. (a) Change from baseline to week 66 in the mNIS+7 composite score, and (b) Change from baseline to week 66 in the Norfolk QoL-DN total score.](/cms/asset/60281c13-94c9-4033-84ae-6e73d4fad96c/ierj_a_1635008_f0005_b.gif)
Significantly greater benefit in secondary and tertiary efficacy endpoints were also shown for inotersen versus placebo [Citation50]. Inotersen was associated with a significant benefit over placebo for all components and subcomponents of the mNIS+7 composite score (all p < 0.05), except for the touch-pressure (p = 0.09) and heart rate to deep breathing (HRDB; p = 0.376) subcomponents of the modified +7 component. The lack of statistically significant effects on touch-pressure and HRDB may be attributable to the ‘ceiling effect,’ in that patients were at the lowest level of the scoring range at baseline; therefore, measurement of change from baseline was difficult. Inotersen was also associated with significant improvements over placebo for all domains of the Norfolk QoL-DN total score (all p ≤ 0.001), except for the small fiber (p = 0.824) and autonomic (p = 0.134) domains. Furthermore, the improvement from baseline to week 65 in the SF-36 physical component summary score was significantly greater with inotersen (LSM difference, 3.59; 95% CI, 1.07 to 6.12; p = 0.006).
Results from the ongoing NEURO-TTR open-label extension (OLE) study (NCT02175004) were presented at the 2018 American Society of Hematology Annual Meeting [Citation53]. Patients from the 15-month NEURO-TTR primary trial who completed study treatment were eligible to enter the OLE study and receive inotersen for up to 5 years (). A total of 135 patients were enrolled in the OLE study. In the OLE, patients in the inotersen group continued to receive inotersen (inotersen–inotersen group) and patients in the placebo group switched to inotersen (placebo–inotersen group). Baseline demographics and disease characteristics were generally well-balanced between the OLE treatment groups. At 2 years of follow-up, patients who switched from placebo to inotersen demonstrated improvement in neuropathy-related quality of life (measured by Norfolk QoL-DN score) and neuropathic progression (mNIS+7) from OLE baseline to week 104 compared with predicted worsening with placebo. Patients who initiated inotersen earlier (i.e. the inotersen–inotersen group) showed greater improvements in neuropathic progression and measures of quality of life than those who switched from placebo to inotersen.
4.3. Investigator-initiated trial
A single-center, investigator-initiated, open-label phase 2 study was conducted to evaluate inotersen in biopsy-proven hereditary or wild-type ATTR patients with moderate-to-severe cardiomyopathy [Citation54]. Subjects were treated with SC inotersen 300 mg, administered once weekly for up to 3 years. To date, 22 patients have been enrolled and 15 have received inotersen for 12 months (ATTRv, n = 8; wild-type ATTR, n = 7).
Of the 15 patients who had received inotersen for 12 months, 8 had ATTRv and 7 had wild-type ATTR. Most patients achieved a maximum reduction of serum TTR levels by week 26 of inotersen therapy, with peak percentage reductions ranging from 39% to 91%. Cardiac structural parameters improved from baseline after 12 months’ treatment with inotersen. In most patients, improvements from baseline in interventricular septal thickness and global systolic strain were observed at 6 and 12 months, with all patients demonstrating a lack of disease progression. In addition, the left ventricular mass measured by magnetic resonance imaging in 10 patients showed a mean reduction of 2.4% at 12 months.
Treatment with inotersen was also associated with stabilization of functional cardiac parameters at 12 months. A mean improvement in the 6-min walk distance test of 29.2 m was observed in the 14 patients who completed this test. Brain natriuretic peptide levels decreased in most patients with ATTRv (5/8 patients) but increased in most wild-type ATTR patients (5/7 patients). Furthermore, New York Heart Association class improved in six patients, remained stable in six patients, and declined in three patients (all three had wild-type ATTR).
This investigator-initiated phase 2 study demonstrated that treatment with inotersen was associated with stabilization of disease progression after 12 months of treatment in patients with ATTR and moderate-to-severe cardiomyopathy. These preliminary findings are encouraging given that significant disease progression over a 12-month period has been previously observed in patients with ATTRv cardiomyopathy [Citation55].
5. Safety and tolerability
In the investigator-initiated phase 2 trial, inotersen was well tolerated by all 15 patients who had received 12 months of inotersen [Citation54]. AEs included pruritus (n = 2), localized injection site reactions (erythema, itching, and induration; n = 5), and transient fatigue (n = 2). Additionally, results of liver function tests, urine analysis, and complete blood count remained stable during the 12 months of inotersen treatment. Although platelet levels decreased by a mean of 11%, no clinically significant bleeding was reported.
During the primary 15-month treatment period of the NEURO-TTR trial, treatment-emergent AEs occurring in ≥10% of patients that were more common in the inotersen group versus the placebo group included nausea (31.3% vs. 11.7%), fatigue (25.0% vs. 20.0%), diarrhea (24.1% vs. 20.0%), headache (23.2% vs. 11.7%), pyrexia (19.6% vs. 8.3%), peripheral edema (18.8% vs. 10.0%), chills (17.9% vs. 3.3%), myalgia (15.2% vs. 10.0%), vomiting (15.2% vs. 5.0%), anemia (13.4% vs. 3.3%), constipation (13.4% vs. 10.0%), thrombocytopenia (13.4% vs. 1.7%), arthralgia (11.6% vs. 8.3%), and decreased platelet count (10.7% vs. 0%) [Citation50]. The mean rate of injection site reactions was 1.1% of all injections in the inotersen group (68 events in 36 patients); 97% of events were mild in severity, and no patient discontinued inotersen prematurely because of an injection site reaction. Postbaseline platelet count decreases to <140,000/mm3 were reported in more patients from the inotersen group compared with the placebo group (54% vs. 13%). Thrombocytopenia with platelet counts of <25,000/mm3 was reported in three (3%) patients in the inotersen group; counts in two of these patients returned to baseline or near-baseline levels after discontinuation of inotersen and treatment with glucocorticoids; in the third case, the patient died of an intracranial hemorrhage before treatment could be initiated. Glomerulonephritis was reported in three (3%) patients in the inotersen group, displaying complex pathologic features on biopsy in all three cases and declines in estimated glomerular filtration rate in two cases. Two out of the three patients with glomerulonephritis were treated with glucocorticoids and regained renal function/improved protein excretion, while permanent hemodialysis was required in one patient. After the implementation of enhanced monitoring, there were no additional cases of severe thrombocytopenia, and a single case of glomerulonephritis was identified early and did not result in loss of renal function. All five deaths reported during the study were in the inotersen group. Four deaths were consistent with progression or complication of the underlying disease. As noted above, the fifth death occurred as a result of a fatal intracranial hemorrhage, which was associated with a platelet count of <10,000/mm3 that occurred before the introduction of weekly platelet monitoring. Other safety variables (vital signs, body weight changes, corrected QT interval, etc.) were stable in both treatment groups, and no clinical manifestations of vitamin A deficiency were reported.
In the NEURO-TTR OLE study, no new safety concerns were observed after 2 years of follow-up [Citation53]. The most common AEs across both treatment groups in the OLE study were similar to those reported in the primary NEURO-TTR trial; these were nausea, urinary tract infection, vomiting, diarrhea, fatigue, chills, fall, peripheral edema, injection site pain, thrombocytopenia, syncope, and injection site erythema. Few patients discontinued treatment with inotersen because of AEs during the OLE study (19/135 patients). Most important, patients appeared to cope well with platelet and renal monitoring during long-term treatment, with no cases of grade 4 platelet count decreases or glomerulonephritis reported during the OLE study.
6. Regulatory affairs
Inotersen received its first global approval on 6 July 2018, in the European Union for the treatment of stage 1 or 2 polyneuropathy in adults with ATTRv. On 3 October 2018, inotersen was also approved in Canada as the first-in-class treatment of stage 1 or stage 2 polyneuropathy in adult patients with ATTRv. On 5 October 2018, the FDA approved inotersen for the treatment of the polyneuropathy of ATTRv in adult patients. Inotersen is administered via a single SC injection at a recommended once-weekly dose of 284 mg, which is equivalent to 300 mg inotersen sodium salt (i.e. the dose used in the NEURO-TTR trial).
The US approval of inotersen includes a black box warning that highlights the increased risk of thrombocytopenia and glomerulonephritis with inotersen therapy. As a result, testing before treatment and monitoring during treatment is required. Because of these increased risks, inotersen is only available through a restricted distribution program called the Tegsedi Risk Evaluation and Mitigation Strategy (REMS) program, which is a strategy employed to manage known or potential risks associated with inotersen to ensure the benefits of the drug outweigh its risks.
7. Conclusion
Inotersen is a novel ASO inhibitor that was developed and approved to reduce the production of TTR in patients with ATTRv. Clinical studies showed that weekly SC injections of inotersen provide significant improvements in neuropathy and QoL, which was sustained during long-term follow-up. In addition, inotersen is generally well tolerated with a manageable safety profile, and regular monitoring for thrombocytopenia and glomerulonephritis has been effective based on results from the OLE study.
8. Expert opinion
Inotersen is a first-in-class therapeutic that targets the underlying pathophysiology of ATTRv offering patients a convenient, once-weekly, self-administered treatment option that improves QoL. Positive improvements in QoL and neuropathy progression were observed in the 15-month NEURO-TTR trial, along with sustained benefits during long-term follow-up in the NEURO-TTR OLE study. Inotersen is also well tolerated, with a manageable safety profile through regular monitoring for the development of glomerulonephritis or thrombocytopenia. Taken together, these findings suggest that all patients with ATTRv who are eligible for treatment with inotersen should initiate treatment early to achieve optimal outcomes.
Genetic testing and ongoing clinical surveillance may result in early detection of asymptomatic carriers, who can then be followed carefully, thus allowing for treatment to be initiated as soon as appropriate before the development of substantial and irreversible damage [Citation56]. In 2016, recommendations made by the European network for TTR-FAP indicated that genetic counseling and routine monitoring for asymptomatic carriers should be undertaken to ensure early detection, thus improving the prognosis of the disease [Citation57]. However, the treatment of asymptomatic patients should not be considered as the penetrance of a mutation may vary among patients, in that symptoms may develop differently in patients with the same mutation [Citation8,Citation56,Citation57]. Indeed, patients without symptoms of neuropathy were not enrolled in the NEURO-TTR or NEURO-TTR OLE study, highlighting the need for studies evaluating the effects of early treatment of patients who do not yet display major symptoms, such as neuropathy and/or cardiomyopathy.
As noted earlier in this article, recent findings of the phase 3 ATTR-ACT trial [Citation31] resulted in the TTR stabilizer, tafamidis, being granted fast-track designation by the FDA and was recently approved for the treatment of cardiomyopathy-associated ATTR. However, the role of inotersen and patisiran in the management of cardiomyopathy-associated ATTRv has not been evaluated in a randomized controlled trial. In the NEURO-TTR trial, a substantial proportion of ATTRv patients had concomitant polyneuropathy and cardiomyopathy [Citation52]. Inotersen has also demonstrated preliminary evidence of efficacy in patients with ATTR cardiomyopathy (both ATTRv and wild-type ATTR) [Citation54]. Therefore, an adequately powered, controlled clinical trial is needed to assess the benefit of inotersen in patients with ATTRv cardiomyopathy.
The introduction of disease-modifying therapies, such as OLT and TTR stabilizers (e.g. tafamidis), has improved survival in patients with ATTRv [Citation31,Citation58,Citation59]. These treatments have altered the natural history of ATTRv, increasing the life expectancy of patients affected by this disease. However, the prolonged disease duration may allow for the disease to progress, resulting in the development of central nervous system (CNS) and ocular manifestations due to the deposition of amyloid fibrils in the brain and eyes. In patients with ATTRv and the Val30Met mutation, previous studies have shown that both CNS and ocular manifestations are associated with a longer disease duration [Citation21,Citation60]. As a result, prospective studies are needed to assess the management of patients with ATTRv who display CNS and/or ocular manifestations.
New TTR-lowering treatment approaches are currently being developed. A next-generation, potent ligand conjugated antisense (LICA) drug (Akcea-TTR-LRx) is currently undergoing clinical development to treat patients with all forms of the disease. Monthly or less frequent SC injections at very low doses may be possible with this novel treatment, which will make this drug an attractive therapeutic option with the potential of an improved safety profile. Furthermore, the design of this next-generation agent should reduce the risk of thrombocytopenia and glomerulonephritis, thereby reducing the need for serial monitoring and potentially decreasing the use of liver transplantation in management. Combination therapy with inotersen and TTR stabilizers is another novel treatment approach that may yield additional benefits in patients with ATTRv, but this approach has not been examined clinically. As is the case with rare disease indications, such as ATTRv, a common limitation is the small patient population for this disease, which prevents adequately powered studies from being conducted. However, the NEURO-TTR trial has demonstrated the benefit of inotersen in ATTRv, and complete follow-up of the NEURO-TTR OLE study through 5 years will provide important long-term efficacy and safety data for inotersen. In addition, exploring the role of RNA-targeted therapies that focus on the disease-forming protein, TTR, in patients with cardiomyopathy without polyneuropathy, will provide valuable information on the role of these agents in the cardiac forms of ATTR amyloidosis. Thus, we predict that the utilization of liver transplantation globally will decline.
Article highlights
Chemical property: molecular formula C230H299N69Na19O121P19S19, molecular weight 7600.73 Da
Mechanism of action and indication: RNA interference drug indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis (ATTRv)
Pharmacokinetics: median time to peak plasma concentration is 2–4 hours, volume of distribution is 293 l, elimination half-life is approximately 1 month
Route of administration and dose: subcutaneous injection dosed at 284 mg (equivalent to 300 mg inotersen sodium salt) once weekly
Most frequent adverse events: nausea, fatigue, diarrhea, headache
Clinical trials: phase 1: healthy volunteers; phase 3: NEURO-TTR trial (NCT01737398; stage 1 or 2 ATTRv with polyneuropathy), NEURO-TTR OLE trial (NCT02175004); investigator-initiated phase 2 trial in hereditary or wild-type transthyretin-mediated amyloidosis with moderate-to-severe cardiomyopathy
Declaration of interest
MA Gertz reports personal fees from Ionis/Akcea, Alnylam, Prothena, Celgene, Janssen, Spectrum, Annexon, Appellis, Amgen, Medscape, Physicians Education Resource, Abbvie (Data Safety Monitoring board), Research to Practice; speaker fees from Teva, Johnson and Johnson, Medscape, DAVA oncology; Advisory Boards for Pharmacyclics and Proclara; Royalties from Springer Publishing; and Grant Funding from Amyloidosis Foundation, International Waldenstrom Foundation, and Spectrum. M Scheinberg has been a consultant and speaker for Pfizer, GSK, Janssen, Roche, Samsung Bioepis, Boehringer Ingelheim, and Novartis. M Waddington-Cruz received honoraria from NHI, Prothena, FoldRx, Ionis, Pfizer, Alnylam, PTC, and Genzyme for travel expenses related to presentations at medical meetings, for acting as a principal investigator in clinical trials and as a consultant member. SB Heitner reports research grants from Pfizer, Eidos, and Ionis and has received consulting fees from Pfizer and Alnylam, Ionis, and Eidos. C Karam has served as a paid consultant for Akcea, Alnylam, Alexion, Biogen, CSL Behring, Cytokinetics, and Genzyme. B Drachman has served on an advisory board for Alnylam Pharmaceuticals; as a study investigator for Ionis Pharmaceuticals, Inc, Alnylam Pharmaceuticals, and Pfizer. S Khella has received honoraria from Akcea Therapeutics and Alnylam Pharmaceuticals. C Whelan has received honoraria from Akcea and Alnylam. L Obici has received speaker fees and participated on scientific advisory boards for Alnylam Pharmaceuticals, Akcea, and Pfizer. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose. Akcea Therapeutics provided a scientific accuracy review at the request of the journal editor.
Additional information
Funding
Related Research Data
References
- Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31.
- Coelho T, Ericzon BG, Falk R, et al. A guide to transthyretin amyloidosis. Clarkston, MI: Amyloidosis Foundation; 2016 cited 2019 Mar 27. http://www.amyloidosis.org/wp-content/uploads/2017/05/2017-ATTR-guide.pdf
- Galant NJ, Westermark P, Higaki JN, et al. Transthyretin amyloidosis: an under-recognized neuropathy and cardiomyopathy. Clin Sci (Lond). 2017;131(5):395−409.
- Obici L. Becoming familiar with hereditary transthyretin amyloidosis, a treatable neuropathy. Arq Neuropsiquiatr. 2018;76(9):573–574.
- Gertz MA. Hereditary attr amyloidosis: burden of illness and diagnostic challenges. Am J Manag Care. 2017;23(7 Suppl):S107–s112.
- Conceicao I, Gonzalez-Duarte A, Obici L, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9.
- Donnelly JP, Hanna M. Cardiac amyloidosis: an update on diagnosis and treatment. Cleve Clin J Med. 2017;84(12 Suppl 3):12–26.
- Hawkins PN, Ando Y, Dispenzeri A, et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625–638.
- Adams D, Coelho T, Obici L, et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015;85(8):675–682.
- Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep. 2014;11(1):50–57.
- Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829–837.
- Liz MA, Mar FM, Franquinho F, et al. Aboard transthyretin: from transport to cleavage. IUBMB Life. 2010;62(6):429–435.
- Rowczenio DM, Noor I, Gillmore JD, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35(9):E2403–E2412.
- Coelho T, Maurer MS, Suhr OB. Thaos - the transthyretin amyloidosis outcomes survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63–76.
- Center for Drug Evaluation and Research. Onpattro 2 mg/mL injection for intravenous use. [cited 2019 Mar 27]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210922Orig1s000Approv.pdf
- European Medicines Agency. Onpattro (patisiran): an overview of Onpattro and why it is authorised in the EU. [cited 2019 Mar 27]. https://www.ema.europa.eu/documents/overview/onpattro-epar-medicine-overview_en.pdf
- Center for Drug Evaluation and Research. Tegsedi injection, 189 mg/mL. [cited 2019 Mar 27]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/211172Orig1s000Approv.pdf
- European Medicines Agency. Tegsedi (inotersen): an overview of Tegsedi and why it is authorised in the EU. [cited 2019 Mar 27]. https://www.ema.europa.eu/documents/overview/tegsedi-epar-summary-public_en.pdf
- Holmgren G, Ericzon BG, Groth CG, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341(8853):1113–1116.
- Adams D, Samuel D, Goulon-Goeau C, et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000;123(Pt 7):1495–1504.
- Maia LF, Magalhaes R, Freitas J, et al. CNS involvement in V30M transthyretin amyloidosis: clinical, neuropathological and biochemical findings. J Neurol Neurosurg Psychiatry. 2015;86(2):159–167.
- Davis MK, Kale P, Liedtke M, et al. Outcomes after heart transplantation for amyloid cardiomyopathy in the modern era. Am J Transplant. 2015;15(3):650–658.
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658–2667.
- Said G, Grippon S, Kirkpatrick P. Tafamidis. Nat Rev Drug Discov. 2012;11(3):185–186.
- Vyndaqel [summary of product characteristics]. Bruxelles, Belgium: Pfizer Europe MA EEIG; 2018.
- Kristen AV, Ajroud-Driss S, Conceicao I, et al. Patisiran, an rnai therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener Dis Manag. 2019;9(1):5–23.
- Coelho T, Maia LF, Da SA M, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792.
- Barroso FA, Judge DP, Ebede B, et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid. 2017;24(3):194–204.
- Keohane D, Schwartz J, Gundapaneni B, et al. Tafamidis delays disease progression in patients with early stage transthyretin familial amyloid polyneuropathy: additional supportive analyses from the pivotal trial. Amyloid. 2017;24(1):30–36.
- Mundayat R, Stewart M, Alvir J, et al. Positive effectiveness of tafamidis in delaying disease progression in transthyretin familial amyloid polyneuropathy up to 2 years: an analysis from the transthyretin amyloidosis outcomes survey (THAOS). Neurol Ther. 2018;7(1):87–101.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016.
- Vyndaqel [prescribing information]. New York, NY: Pfizer Labs; 2019 May.
- Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21.
- Verona G, Mangione PP, Raimondi S, et al. Inhibition of the mechano-enzymatic amyloidogenesis of transthyretin: role of ligand affinity, binding cooperativity and occupancy of the inner channel. Sci Rep. 2017;7(1):182.
- Sant’Anna R, Gallego P, Robinson LZ, et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat Commun. 2016;7:10787.
- Kolstoe SE, Mangione PP, Bellotti V, et al. Trapping of palindromic ligands within native transthyretin prevents amyloid formation. Proc Natl Acad Sci U S A. 2010;107(47):20483–20488.
- Mu Y, Jin S, Shen J, et al. CHF5074 (CSP-1103) stabilizes human transthyretin in mice humanized at the transthyretin and retinol-binding protein loci. FEBS Lett. 2015;589(7):849–856.
- Qiang L, Guan Y, Li X, et al. CSP-1103 (CHF5074) stabilizes human transthyretin in healthy human subjects. Amyloid. 2017;24(1):42–51.
- Miller M, Pal A, Albusairi W, et al. Enthalpy-driven stabilization of transthyretin by AG10 mimics a naturally occurring genetic variant that protects from transthyretin amyloidosis. J Med Chem. 2018;61(17):7862–7876.
- Penchala SC, Connelly S, Wang Y, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci U S A. 2013;110(24):9992–9997.
- Cardoso I, Martins D, Ribeiro T, et al. Synergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010;8:74.
- Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012;19(Suppl 1):34–36.
- Richards DB, Cookson LM, Berges AC, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med. 2015;373(12):1106–1114.
- Higaki JN, Chakrabartty A, Galant NJ, et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of transthyretin. Amyloid. 2016;23(2):86–97.
- Galant NJ, Bugyei-Twum A, Rakhit R, et al. Substoichiometric inhibition of transthyretin misfolding by immune-targeting sparsely populated misfolding intermediates: a potential diagnostic and therapeutic for TTR amyloidoses. Sci Rep. 2016;6:25080.
- Benson MD, Dasgupta NR, Monia BP. Inotersen (transthyretin-specific antisense oligonucleotide) for treatment of transthyretin amyloidosis. Neurodegener Dis Manag. 2019;9(1):25–30.
- Tegsedi [prescribing information]. Boston, MA: Akcea Therapeutics;October2018
- Niemietz CJ, Sauer V, Stella J, et al. Evaluation of therapeutic oligonucleotides for familial amyloid polyneuropathy in patient-derived hepatocyte-like cells. PLoS One. 2016;11(9):e0161455.
- Ackermann EJ, Guo S, Benson MD, et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-generation antisense oligonucleotides. Amyloid. 2016;23(3):148–157.
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31.
- Tegsedi [summary of product characteristics]. London, UK: Akcea Therapeutics UK Ltd.; 2018.
- Waddington-Cruz M, Ackermann EJ, Polydefkis M, et al. Hereditary transthyretin amyloidosis: baseline characteristics of patients in the NEURO-TTR trial. Amyloid. 2018;25(3):180–188.
- Brannagan T, Wang AK, Coelho T, et al. Long-term update from the open-label extension of the NEURO-TTR study in patients with hereditary transthyretin amyloidosis. Presented at: American Society of Hematology Annual Meeting and Exposition; December 1–4, 2018; San Diego, CA.
- Benson MD, Dasgupta NR, Rissing SM, et al. Safety and efficacy of a TTR specific antisense oligonucleotide in patients with transthyretin amyloid cardiomyopathy. Amyloid. 2017;24(4):219–225.
- Benson MD, Teague SD, Kovacs R, et al. Rate of progression of transthyretin amyloidosis. Am J Cardiol. 2011;108(2):285–289.
- Schmidt HH, Barroso F, Gonzalez-Duarte A, et al. Management of asymptomatic gene carriers of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2016;54(3):353–360.
- Obici L, Kuks JB, Buades J, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016;29(Suppl 1):S27–35.
- Ericzon BG, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–1854.
- Rosenblum H, Castano A, Alvarez J, et al. (transthyretin) stabilizers are associated with improved survival in patients with TTR cardiac amyloidosis. Circ Heart Fail. 2018;11(4):e004769.
- Beirao JM, Malheiro J, Lemos C, et al. Ophthalmological manifestations in hereditary transthyretin (ATTR V30M) carriers: a review of 513 cases. Amyloid. 2015;22(2):117–122.