
ABSTRACT
Introduction: The proven efficacy of the cellular vaccine sipuleucel-T in 2010 led to optimism about immunotherapeutic approaches for the treatment of prostate cancer. Some surmised that prostate cancer might be an ideal target for immune-mediated killing given that the prostate is not an essential organ and expresses unique proteins including prostate-specific antigen, prostate-specific membrane antigen, and prostatic acid phosphatase that could be targeted without side effects. Subsequently, antibodies that inhibit the T cell checkpoints PD1 and CTLA4 were shown to stimulate antitumor immune responses, leading to tumor regression in several cancer types. These therapies have since been tested in several studies as treatments for prostate cancer, but appear to have limited efficacy in molecularly unselected patients.
Areas covered: In this review, we discuss these studies and evaluate features of prostate cancer and its host environment that may render it generally resistant to CTLA4 and PD1 blockade. We provide an overview of alternate immune checkpoints that may hold greater significance in this disease.
Expert opinion: Combination therapies to target multiple layers of alternate immune checkpoints may be required for an effective immune response to prostate cancer. We discuss combination therapies currently being investigated.
1. Introduction
Historically, cancer drug development efforts have focused on directly inhibiting pathways of cancer cell growth and survival. In contrast, cancer immunotherapy aims to stimulate an antitumor immune response. Tumor immunity is theoretically superior to pharmacologic tumor inhibition because it can be targeted, adaptable, and persistent over time. To date, the most successful strategy to induce immunity to solid tumors is inhibition of T cell co-inhibitory pathways, i.e. T cell checkpoints[Citation1]. T cell activation occurs following ligation of the T cell receptor (TCR) and the co-stimulatory molecule CD28, which leads to upregulation of co-inhibitory molecules including CTLA4, PD1, LAG-3, TIGIT, and TIM-3 [Citation2]. Activation of these inhibitory pathways subsequently restrains T cells to elegantly balance protection from pathogens with collateral damage to the host and induction of autoimmunity. Inhibition of these co-inhibitory pathways by T cell immune checkpoint inhibitors tilts this balance toward enhanced T cell pro-inflammatory function and augmentation of antitumor immune responses [Citation3]. Moreover, some tumors learn to evade the immune system by activating T cell checkpoints. Together, these phenomena have led to the development of inhibitors of CTLA4 and PD1/PDL1 as therapeutic agents for cancer.
Significant effort has been placed into determining whether there might be a role for these T cell checkpoint inhibitors in treatment of patients with prostate cancer. Unfortunately, these agents appear to have limited efficacy in unselected patients. Here, we review the biological effects of inhibition of the T cell checkpoints CTLA4 and PD1, the results of studies that have tested these agents in prostate cancer, and a framework to consider alternate immune checkpoints in this disease.
2. Biological effect of CTLA4 blockade
CTLA4 is required for maintenance of immune tolerance. Mice with genetic deletion of CTLA4 exhibit fatal systemic inflammation [Citation4,Citation5]. Similarly, humans with genetic disorders of CTLA4 deficiency (CTLA4 Haploinsufficiency with Autoimmune Infiltration [CHAI] disease, and LRBA deficiency with Autoantibodies, Treg defects, Autoimmune Infiltration, and Enteropathy [LATAIE] disease) exhibit immune dysregulation with autoimmune features [Citation6]. CTLA4 restrains autoimmunity through direct inhibition of effector T cell function and indirect inhibition via activation of regulatory T cells (Tregs) [Citation7]. Effector T cell stimulation leads to rapid upregulation of CTLA4 [Citation8], which subsequently can inhibit T cell activation. Mechanistically, CTLA4 may both inhibit intracellular signaling downstream of the TCR and CD28 [Citation9], as well as compete for binding of B7-1 and B7-2, sequestering them away from binding to CD28 and leading to loss of effective co-stimulation [Citation10]. Tregs constitutively express CTLA4, which is required for their effective immune suppressive function [Citation11,Citation12].
It was first shown that CTLA4 inhibition can augment antitumor immune responses in a seminal study from the laboratory of James Allison that showed that treatment with anti-CTLA4 antibodies can induce immunity to tumors in mice [Citation3]. Subsequent work indicated that both effector and regulatory T cells are important cellular targets of CTLA4 inhibition [Citation13,] (). CTLA4 inhibition of each cell type alone showed modest effect on antitumor immunity, while targeting of both compartments led to an additive effect . Anti-CTLA4 can lead to expansion of tumor antigen-specific effector T cells [Citation14–16] and alterations to the composition of T lymphocyte populations within tumors [Citation17]. Specifically, tumors regressing due to anti-CTLA4 showed expansion of exhausted-like CD8 T cells, enrichment of an ICOS+Th1-like CD4 effector population, and depletion of Tregs. Depletion of Tregs appears to be driven by antibody-dependent cellular cytotoxicity by FcɣR-positive macrophages within the tumor [Citation18].
Figure 1. Simplified schematic of the biological effect of CTLA4 and PD1 blockade. antibodies targeting CTLA4 remove negative regulation of T cell priming in the lymph node and can lead to depletion of tregs in tumors. antibodies targeting the PD1 pathway remove negative regulation of T cell activation in tumors and can lead to depletion of tregs and MDSCs. (Adapted from ‘Blockade of CTLA-4 or PD-1 signaling in tumor immunotherapy,’ by BioRender.com (2021). retrieved from https://app.biorender.com/biorender-templates)
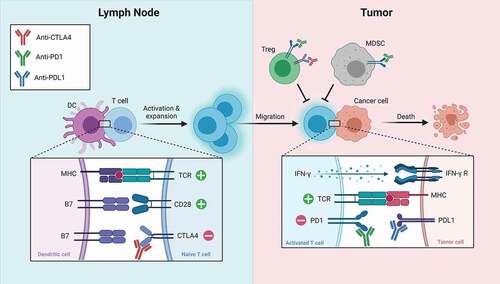
Two human monoclonal antibodies to CTLA4 have been developed for clinical use, ipilimumab and tremelimumab, although only ipilimumab has been approved by the US Food and Drug Administration (FDA; 2011). Patients treated with anti-CTLA4 may mimic some features of CHAI and LATAIE disease, including propensity to nonspecific autoimmunity. This is a significant limitation of this therapy and supports the concept that the mechanism of action of this drug is global dysregulation of immune tolerance that incidentally may lead to an antitumor immune response. Efforts are ongoing to make anti-CTLA4 therapy more directed toward generating a specific antitumor response by targeting the drug to the tumor microenvironment [Citation19].
3. Biological effect of PD1 blockade
PD1 maintains immune tolerance by a distinct mechanism from CTLA4. Although both co-inhibitory molecules are expressed by T cells following activation, their ligands are expressed by different cell types that are encountered by the T cell at different stages of activation, expansion, and differentiation, and usually in different anatomical locations [Citation20,Citation21]. The ligands for CTLA4, B7-1 and B7-2, are expressed by antigen-presenting cells (APCs) and thus are encountered early during T cell priming, which is thought to occur primarily in secondary lymphoid structures. In contrast, the ligands for PD1, PD-L1 and PD-L2, are expressed by non-lymphoid tissue [Citation22], and are encountered late when expanded cells are exerting effector function in the periphery (). PD1 ligation leads to inhibition of signaling downstream of the TCR [Citation23,Citation24], pro-inflammatory cytokine production [Citation25], and metabolic reprogramming required for a pro-inflammatory response [Citation26,Citation27]. Additionally, PD1 signaling can trigger production and maintenance of inducible regulatory T cells [Citation28]. Moreover, unlike CTLA-4, PD1 is also expressed on B cells, natural killer (NK) cells, and myeloid-derived suppressor cells (MDSCs) [Citation29], where it also appears to suppress an immune response. Notably, autoimmunity of PD1-deficient mice is less severe than CTLA4-deficient mice, and the specific type of autoimmunity depends on the genetic background of the mouse [Citation30–32].
Thus, inhibition of PD1 is not equivalent to inhibition of CTLA4. There are currently six FDA-approved human monoclonal antibodies that inhibit PD1 signaling: nivolumab, pembrolizumab, and cemiplimab (which inhibit PD1); and atezolizumab, avelumab, and durvalumab (which inhibit PD-L1). Two non-FDA-approved anti-PD1 agents in clinical trials for patients with prostate cancer include zimberelimab and MGA012. Similar to mouse models, patients treated with these agents generally exhibit fewer and less severe immune toxicities than patients treated with ipilimumab. Moreover, tumors regressing on anti-PD1 showed expansion of exhausted-like CD8 T cells similar to anti-CTLA4, however distinct from anti-CTLA4-treated tumors, there was no alteration in the CD4 T cell compartment and the magnitude of Treg depletion was less in anti-PD1-treated tumors. Given their complementary and perhaps sequential effects, inhibition of PD1 and CTLA4 concurrently can have additive antitumor effects. Notably, the ultimate mechanism of T cell-mediated killing of tumor cells stimulated by CTLA4 blockade and PD1 blockade appears to be similar and require IFNɣ-signaling within tumors [Citation33–35].
4. Efficacy of CTLA4 blockade in prostate cancer
The clinical efficacy of CTLA4 blockade in advanced prostate cancer was rigorously assessed in the CA184-043 study, a randomized, double-blind, phase 3 trial of ipilimumab versus placebo following one dose of bone-directed radiotherapy for 799 patients with metastatic castration-resistant prostate cancer (mCRPC) that has progressed on docetaxel [Citation36]. This trial failed to meet its primary endpoint, and did not show a statistically significant improvement in median overall survival (OS) (11.2 months for ipilimumab versus 10.0 months for placebo). However, the hazard ratio for death was noted to decrease over time; while it was 1.46 (95% CI 1.10–1.95) for 0–5 months, it decreased to 0.65 (0.50–0.85) for 5–12 months, and 0.60 (0.43–0.86) beyond 12 months. An updated analysis of this study with 2.4 years of additional follow-up confirmed this finding that ipilimumab potentially conferred a survival benefit at late time points [Citation37]. The most plausible explanation for this finding is that a small subset of patients derived significant benefit from ipilimumab. This patient population was small and therefore only became visible after the majority of patients (who did not benefit) had died. Immune-related adverse events (irAEs) occurred in 63% of patients treated with ipilimumab and 22% of patients treated with placebo; 26% of patients in the ipilimumab group experienced grade 3–4 irAE, and 89% of irAEs resolved after standard irAE management algorithms were followed.
The investigators did make an effort to identify biomarkers of sensitivity within the CA184-043 study, however tumor DNA sequencing was not performed. There was a suggestion that patients with favorable disease characteristics including those without visceral metastases and lower tumor marker levels may have derived more benefit from ipilimumab. This garnered anticipation for results of the subsequent CA184-095 trial, which was a randomized, double-blind phase 3 trial of ipilimumab versus placebo for 602 patients with chemotherapy-naïve mCRPC [Citation38]. In that trial, ipilimumab was associated with a longer median progression-free survival (PFS) (5.6 versus 3.8 months), longer time to chemotherapy administration, and a higher PSA response (23% versus 8%) than placebo. Yet unfortunately, this study also failed to show a survival benefit to treatment with ipilimumab, and unlike CA184-043, there was no late separation of the Kaplan-Meier OS curves. Discordance between PFS and OS in this trial suggests that patients may have experienced adverse effects of ipilimumab that nullified initial antitumor benefit, or that PFS is not a good surrogate of OS in this disease context. In this trial, treatment-related AEs occurred in 82% of patients treated with ipilimumab and 49% of patients treated with placebo; grade 3–4 treatment-related AEs occurred in 40% and 6% of each group, respectively.
These two large phase 3 trials indicate that ipilimumab as a single agent is ineffective for the vast majority of patients with mCRPC, and this drug has not been FDA-approved for use in prostate cancer. Yet the question remains why CA184-043 demonstrated late separation of the survival curves, while CA184-095 did not. The two major differences in these trials were the administration of bone-directed radiotherapy and prior treatment with chemotherapy. This may suggest that there is a small subset of patients with mCRPC who are sensitive to ipilimumab, but only following treatment with radiotherapy or chemotherapy or both.
5. Efficacy of PD1 blockade in prostate cancer
The initial pan-cancer phase 1 study of nivolumab included 17 patients with CRPC; however, none experienced an objective response [Citation39]. Subsequent early studies with pembrolizumab indicated that patients with CRPC are not uniformly resistant to PD1 blockade, and identified some patients who experienced significant benefit [Citation40,Citation41]. This possibility was further explored in KEYNOTE-199, which is a large open-label, phase 2 trial of pembrolizumab used alone (cohorts 1–3, for mCRPC that has progressed on chemotherapy) or in combination with ongoing enzalutamide (cohorts 4–5, for chemotherapy-naïve mCRPC that has progressed on enzalutamide) [Citation42]. The results of cohorts 1–3 have been published. Cohort 1 included 133 patients with PD-L1-positive measurable disease and exhibited an objective response rate (ORR) of 5%, median PFS of 2.1 months, and median OS of 9.5 months. Cohort 2 included 66 patients with PD-L1-negative measurable disease and exhibited an ORR of 3%, median PFS of 2.1 months, and median OS of 7.9 months. Cohort 3 included 59 patients with bone-predominant disease (irrespective of PD-L1 status) and exhibited a median PFS of 3.7 months and median OS of 14.1 months. Conclusions are limited without control arms; however, the results suggest that this agent was safe, PD-L1 expression is not a robust marker for sensitivity to PD1 blockade in advanced prostate cancer, and that anti-PD1 is not broadly effective as a single agent in these patients. Interestingly, objective response rates and PSA response rates to pembrolizumab tended to be higher in patients with homologous recombination repair (HRR) gene mutations, especially in men with BRCA2 or ATM mutations; this finding remains to be confirmed. Preliminary data from cohorts 4–5 have been presented in the abstract form [Citation43]. These results suggest a slightly higher response rate of pembrolizumab when used in combination with enzalutamide although it is possible this was due to inclusion of patients with chemotherapy-naïve mCRPC. Full immune and genetic biomarker analyses from the KEYNOTE-199 study are eagerly awaited to define the small subset of patients that may have benefited from pembrolizumab treatment.
6. Efficacy of combined PD1 and CTLA4 blockade in prostate cancer
The clinical activity of combined CTLA4 and PD1 blockade was first assessed in a single-arm, open-label, phase 2 study of 15 patients with AR-V7-positive mCRPC treated with nivolumab (3 mg/kg) and ipilimumab (1 mg/kg) for four cycles, followed by maintenance nivolumab [Citation44]. The rationale for this study was that this subset of patients has poor prognosis, limited treatment options, and may be enriched for mutations in HRR genes, which were predicted to enhance response to ICB. An objective response occurred in 2 of 8 patients with measurable disease, the median PFS was 3.7 months, and the median OS was 8.2 months. Patients with HRR defects (especially those with BRCA2 or ATM mutations) had longer median PFS, so this study supports further investigation of combined CTLA4 and PD1 blockade in this subset of patients, but not necessarily in those with AR-V7–positive disease.
A larger trial of combined ipilimumab and nivolumab was subsequently performed, entitled CheckMate 650 [Citation45]. This was an open-label, phase 2 study of nivolumab (1 mg/kg) and ipilimumab (3 mg/kg) for four cycles, followed by maintenance nivolumab for 90 patients with mCRPC. The rationale for this trial was that ipilimumab treatment alone prior to prostatectomy was found to increase expression of PD-L1 and VISTA inhibitory molecules on macrophages [Citation46]. Thus, the authors hypothesized that PD1 may become an important induced immune checkpoint following ipilimumab. Cohort 1 included patients with chemotherapy-naïve mCRPC and exhibited an ORR of 25%, median PFS 5.5 months, and median OS 19 months. Cohort 2 included patients with mCRPC that had progressed on chemotherapy and exhibited an ORR of 10%, median PFS 3.8 months, and median OS 15.2 months. These are the most encouraging metrics of efficacy for ICB in prostate cancer to date, and certainly support further investigation of dual CTLA4 and PD1 checkpoint blockade for this disease. Once again, there was a suggestion that patients with mutations in HRR genes derived slightly greater benefit, and this hypothesis requires formal prospective confirmation.
Yet despite encouraging efficacy results of CheckMate 650, the combination regimen appeared quite toxic as approximately half of the patients experienced grade 3–4 treatment-related adverse events, and 4 patients experienced treatment-related deaths. This is in contrast to the aforementioned study of nivolumab (3 mg/kg) and ipilimumab (1 mg/kg) for patients with AR-V7-positive mCRPC. Although that study was smaller, only 33% of the patients experienced grade 3–4 treatment-related adverse events, and there were no treatment-related deaths. This may suggest that this dosing regimen is safer for the ongoing investigation of combined CTLA4 and PD1 blockade. Alternative dosing and scheduling of the ipilimumab plus nivolumab combination is currently being explored.
7. A working model of efficacy of CTLA4 and PD1 blockade in prostate cancer
Following review of trials of blockade of CTLA4 and PD1 in prostate cancer, it is clear that these agents are not broadly effective for the majority of patients with advanced prostate cancer. However, we do have the optimistic viewpoint that these trials were not entirely negative. In each trial discussed above, there was a signal (albeit small) that some patients obtained a benefit. Needless to say, it is critical to understand who these patients are and why they responded so that we might offer treatment to those most likely to benefit and develop rational strategies to overcome resistance in the remainder.
An important consideration is that CTLA4 and PD1 activation are often not the only barriers between a tumor and an antitumor immune response. Tumor immunity involves several cell types and different anatomical locations, and is proposed to function in a cyclical manner, termed the ‘Cancer-Immunity Cycle,’ with a multitude of stimulatory and inhibitory factors [Citation21]. Key requirements include tumor production of neoantigens, innate immune cell activation and T cell priming, a tumor microenvironment supportive of pro-inflammatory immune cell function, and tumor sensitivity to T cell killing. While CTLA4 and/or PD1 blockade can remove some negative regulation of tumor immunity, it likely cannot compensate for complete lack of one of these key factors. In fact, some argue that these agents are only effective when the infrastructure of an endogenous antitumor immune response is in place, and primed T cells are only restrained by checkpoint activation or similar induction of exhaustion [Citation47]. This notion is supported by evidence that tumors, including prostate tumors, with baseline inflamed gene signatures were more likely to respond favorably to ipilimumab [Citation48,Citation49] and pembrolizumab [Citation50]. Thus, we must consider: what tends to be the endogenous immune response to prostate cancer, and what are the barriers to effective prostate cancer tumor immunity, if not activation of T cell immune checkpoints? The remaining sections of this Review will address these questions.
8. Expert opinion: the endogenous immune response in advanced prostate cancer
Inflammation is purported to initiate prostate carcinogenesis [Citation51]. As such, prostate cancer must learn to cooperate with inflammation from its outset. Primary untreated prostate cancer exhibits remarkable homogeneity in infiltrating immune cell composition, with high relative abundance of follicular helper T cells, eosinophils, and mast cells [Citation52]. In contrast, metastatic (and usually previously hormonally treated) prostate cancers are often described as ‘immunologically cold’ tumors with low densities of tumor-infiltrating lymphocytes (TIL) on the order of 100 CD8 + T cells per mm2 [Citation53–55]. As a point of comparison, metastatic melanomas tend to have TIL densities on the order of 1000 CD8+ cells per mm2, with higher baseline TIL density associating with greater responses to PD-1 blockade [Citation56]. The low density of TILs in the majority of metastatic prostate cancers suggests that this tumor type does not invoke a robust endogenous adaptive immune response. Immunophenotyping of the small proportion of TILs in mCRPC suggests that a large proportion of CD4+ cells are Tregs, and a large proportion of CD8+ cells exhibit a dysfunctional phenotype with expression of PD1, TIM3, TIGIT, ICOS, FASL, and LAG3 [Citation57]. Thus, perhaps inhibiting T cell checkpoints is ineffective because there are so few T cells in the metastatic prostate cancer microenvironment, and those that are present are rendered dysfunctional by mechanisms beyond PD1 and CTLA4 activation. Therefore, perhaps we should look beyond these T cell checkpoints and consider earlier and alternate checkpoints to an endogenous immune response to prostate cancer. Three broad immune checkpoints and strategies to target them will be discussed, including tumor production of neoantigens, innate immune cell function, and T cell function ().
Figure 2. Barriers to prostate cancer immunity. (original figure, created with bioRender.com).
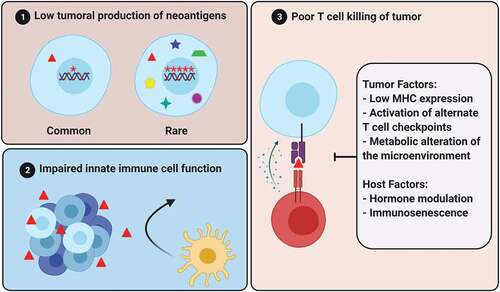
9. Barrier 1: low tumoral production of neoantigens
Antigens are molecules that are recognized and elicit an immune response in the context of inflammation. Cancer antigens are often derived from mutated self-peptides against which the immune system has not been tolerized and are called “neoantigens.” Production and presentation of neoantigens are likely required for a tumor adaptive immune response. Neoantigen burden loosely correlates with tumor mutational burden (TMB), which is an important biomarker of response to ICB [Citation58]. High TMB can be driven by loss of function of one or more of the DNA mismatch repair proteins (MSH2, MSH6, MLH1, and PMS2), i.e. mismatch repair deficiency (dMMR)[Citation59], or by POLE or POLD1 polymerase gene mutations affecting the proofreading/exonuclease domains. Advanced dMMR solid tumors exhibit high response rates to ICI [Citation60] and case reports indicate that solid tumors with POLE or POLD1 mutations (which are much more rare) may behave similarly [Citation61–63]. Both high TMB (>10 mutations per megabase (mut/Mb)) and dMMR are FDA-approved indications for use of ICB in patients with advanced solid tumors, irrespective of histologic type.
Production of neoantigens may be low in the majority of prostate cancers, which may be a significant barrier to response to T cell checkpoint inhibitors. Median TMB of unselected treatment-naïve localized prostate adenocarcinoma ranges from 1 to 2 non-synonymous mut/Mb [Citation64], which is among the lowest of all solid tumor types [Citation65]. The rare subset of patients with mCRPC with dMMR are estimated to have a median TMB of 10–20 mut/Mb and exhibit high PSA response rates to PD1 blockade, in the order of 40–60% [Citation66–70]. CDK12 inactivation has been shown to lead to focal tandem duplications and production of gene fusions that may function as neoantigens [Citation71,Citation72], and patients with mCRPC with CDK12 mutations exhibited higher than expected PSA responses to ICB in small retrospective case studies [Citation73,Citation74]. Similarly, BRCA2 mutations are associated with more indel alterations and expression of potential neoantigens [Citation75], which may in part explain the signal of increased efficacy of ICB among patients with HR mutations as described above. Given that responses to ICB can be elicited when neoantigens are not limiting in prostate cancer, this supports an idea that a major barrier to response to T cell checkpoint inhibitors in unselected prostate cancer is low production of neoantigens.
As such, key areas of future investigation is the identification of markers of high neoantigen production beyond TMB and strategies to induce neoantigen production. It seems that the types of mutations that contribute to TMB may be important as small nucleotide insertion-deletion mutations that lead to frameshifts or gene fusions are more likely to generate peptides that appear foreign and can function as neoantigens compared to single nucleotide variants [Citation76]. For example, while renal cell carcinoma generally exhibits a low TMB, it has a high proportion of indel mutations leading to frameshifts that are associated with favorable response to ICB [Citation77]. In prostate cancer with dMMR, the frameshift mutation proportion has been linked with greater response to PD1 blockade compared to overall TMB [Citation66]. It is not known whether frameshift mutations, which are relatively rare outside of dMMR, might predict the response to ICB in MMR-proficient prostate cancer.
Strategies to induce neoantigen production are sparse. Theoretically, agents that directly damage DNA (e.g., platinum) or inhibit repair of DNA damage (e.g. PARP inhibitors) might increase TMB and neoantigen production over time. Despite the fact that this idea is frequently put forth in the literature, most data suggest these agents only minimally alter TMB [Citation78].
Perhaps a more promising approach is to boost immune responses to weak but ubiquitously expressed endogenous tumor antigens, such as prostatic acid phosphatase (PAP), the androgen receptor (AR) or prostate-specific antigen (PSA), through vaccines (). Sipuleucel-T is an autologous cellular therapy in which patients’ antigen-presenting cells (APCs) are extracted, loaded and stimulated with PAP ex vivo, and re-infused. This leads to antigen-specific T cell activation [Citation79] and immune-cell recruitment into tumor [Citation80] and confers a median 4.1 month increase in OS [Citation81]. Small trials of sipuleucel-T in combination with ipilimumab [Citation82] or pembrolizumab [Citation83] reported encouraging results that may indicate synergy. A larger trial of ipilimumab in combination with sipuleucel-T is ongoing (NCT01804465).
Table 1. Selected therapeutic strategies to target barriers to prostate cancer tumor immunity
DNA vaccines encoding PAP (pTVG-HP) and AR (pTVG-AR) have been shown to be safe, and induce Th1 responses to PAP and AR, respectively [Citation84,Citation85]. Although pTVG-HP did not prolong metastasis-free survival among patients with non-metastatic biochemically recurrent prostate cancer, a trial is underway to evaluate the effect of pTVG-HP with and without pTVG-AR in combination with pembrolizumab in patients with mCRPC (NCT04090528). Similarly, the vaccines PROSTVAC (recombinant poxvirus to express PSA) and W_pro1 (mRNA encoding 5 prostate cancer antigens) are currently in phase 1/2 clinical trials in combination with anti-PD1 agents (NCT02933255 and NCT04382898). Another agent that may direct cytotoxic T cells to prostate cancer in the absence of presented neoantigens may be AMG160, which is a PSMAxCD3 bispecific T cell engager (BiTE), and a phase 1 trial is ongoing to assess the feasibility of combining this agent with pembrolizumab (NCT03792841).
10. Barrier 2: impaired innate immune cell function and poor T cell priming
Yet low tumor production of neoantigen is certainly not the only barrier to an effective and durable antitumor immune response in patients with prostate cancer. This is illustrated by the fact that although patients with mismatch-repair deficient mCRPC exhibit a high response rate to anti-PD1, it is not universal, and the responses seem to be of shorter duration than mismatch-repair deficient cancers of other primary histology [Citation66].
Neoantigens are taken up, processed, and presented by antigen-presenting cells (APCs) such as dendritic cells and tumor-associated macrophages (TAMs), which often must traffic to secondary lymphoid organs to encounter and prime antigen-specific T cells. CD8α+ conventional dendritic cells appear to be the most critical APC for cross-presentation of neoantigen for tumor rejection by T cells [Citation86,Citation87]. APC activation occurs in coordination with other innate immune cells including NK cells, NK T cells, and γδ T cells in response to damage-associated molecular patterns (DAMPs) [Citation88–90]. The function of innate immune cells in the tumor microenvironment is critical to downstream adaptive immune cell function.
Several studies indicate that innate immune cells are dysfunctional in the prostate tumor microenvironment. A high ratio of circulating neutrophils to lymphocytes has been associated with poor prognosis [Citation91,Citation92]. In fact, innate immune cells often seem to be reprogrammed to be immune suppressive (and tumor-promoting), rather than pro-inflammatory. Myeloid-derived suppressor cells (MDSCs) are abundant, and the immune suppressive M2 subtype of TAMs increase in numbers from normal prostate to mCRPC [Citation93]. Myeloid cells may be induced to differentiate to M2 TAMs by tumor-secreted factors in the prostate cancer microenvironment as conditioned media from prostate cancer cell lines (but not colorectal cell lines) was sufficient to drive macrophages into the M2, TGF-β-secreting, subtype [Citation94]. Specifically, CXCL2 was nominated to be a key factor that activates CXCR2 on macrophages and polarizes them toward the M2 lineage [Citation95]. Inhibition of CXCR2 led to increased pro-inflammatory macrophages and restriction of prostate tumor growth in the mouse, and the CXCR2 inhibitor navarixin is currently being studied in a phase 2 trial in combination with pembrolizumab for patients with mCRPC (NCT03473925). Importantly, selective elimination of M2 TAMs suppresses tumor growth, extends survival, and augments the effect of ICB in murine cancer models [Citation96]. In fact, some evidence suggests that the chemotherapy agent docetaxel, which is frequently used for treatment of prostate cancer, may owe some of its efficacy to selective suppression of M2 TAMs [Citation97]. Trials are ongoing to assess the effect of combination therapy with docetaxel and T cell checkpoint inhibitors (NCT03834506, NCT04100018, NCT03879122). The agent BXCL701 is purported to inhibit dipeptidyl peptidases leading to macrophage death, and is also being investigated in combination with pembrolizumab in patients with mCRPC (NCT03910660).
Similarly, the tyrosine kinase inhibitor cabozantinib was shown to deplete M2 TAMs and synergize with T cell checkpoint inhibitors in preclinical models [Citation98]. Interestingly, a separate study suggested that cabozantinib additionally stimulates the release of neutrophil chemotactic factors, CXCL12 and HMGB1, leading to neutrophil-mediated antitumor immune responses and clearance of PTEN/p53-deficient murine prostate cancer [Citation99]. The combination of cabozantinib and atezolizumab was assessed in 44 patients with mCRPC in a phase 1b trial called COSMIC-021 [Citation100]. The results appeared better than should be expected for atezolizumab or cabozantinib alone and reported a PSA response rate of 67% and an ORR of 32% with two complete responses and 12 partial responses. A phase 3 study is underway to look more carefully at this combination (NCT04446117). The mechanism of M2 TAM immune suppression may be the production of TGF-β, which has been described to reprogram peritumoral stroma to exclude CD8+ effector T cells and inhibit an antitumor immune response [Citation101]. Trials of TGF-β receptor inhibition with the drug galunisertib are ongoing for patients with mCRPC (NCT02452008). A recent study of mCRPC found that enzalutamide exposure increased gene expression of TGF-β signaling [Citation57] suggesting that targeting this pathway may become even more important as CRPC acquires resistance to potent AR inhibition. Although it is not innate immune cells, some evidence suggests that tumor-associated plasma cells are associated with higher T cell infiltration into primary prostate cancer and better outcomes after local therapy [Citation102,Citation103]; however, the functional role of this cell type is not clear.
A complementary therapeutic strategy to mobilize an innate immune response in prostate cancer beyond inhibition of M2 TAMs is APC activation. Indeed, the proven efficacy of sipuleucel-T, in which activated APCs loaded with PAP are supplemented, may suggest that APC activation is limiting in the endogenous immune response to prostate cancer. Furthermore, in effective endogenous tumor immune responses, tumor-derived DNA functions as a DAMP to stimulate STING (stimulator of interferon genes) in APCs, which leads to APC activation, cytokine production, and effective T cell priming [Citation104,Citation105]. Intratumoral injection of the STING agonist cyclic di-GMP augmented the therapeutic effect of CTLA4, PD1, and 41-BB inhibition in murine models of prostate cancer [Citation106]. Notably, PARP inhibitors may also lead to high levels of double-stranded DNA fragments that can activate STING-dependent type-1 IFN in APCs. This was first described in BRCA1- or BRCA2-deficient tumors [Citation107,Citation108], but has since been suggested to occur even in tumors with intact BRCA1/2 function [Citation109]. Trials are ongoing to assess the efficacy of combining the PARP inhibitors olaparib or rucaparib with T cell checkpoint inhibitors in unselected mCRPC (NCT03834519, NCT03338790).
Finally, we and others have been investigating the use of cycling high-dose testosterone (called bipolar androgen therapy; BAT) for treatment of advanced prostate cancer [Citation110]. Interestingly, recent studies suggest that supraphysiological levels of androgen may stimulate NF-κB signaling within prostate cancer cells leading to mobilization of cytotoxic T cells into tumors, likely due to innate immune cell activation [Citation54]. To this end, the combination of BAT plus nivolumab is being investigated in a phase 2 trial for patients with mCRPC (NCT03554317; COMBAT). Additional strategies to invigorate an innate immune response to prostate cancer are currently being studied in other clinical trials, which are listed in .
11. Barrier 3: poor T cell killing of tumor cells
Following priming by APCs, neoantigen-specific cytotoxic T cells must traffic to the tumor and kill the tumor cells. For this to occur, healthy tumor-specific T cells must exist, evade T cell checkpoints, recognize tumor, and the tumor must be sensitive to immunogenic cell death. Each of these steps may represent an impediment in prostate cancer immunotherapy and could be considered for therapeutic modulation.
T cell health in patients with prostate cancer. Patients with prostate cancer tend to be of advanced age. For example, the median age of patients in CA184-043 was 68 years, while the median age of patients in the initial trial of ipilimumab for advanced melanoma was 56 years [Citation111]. Lymphocytes from older hosts may exhibit dysfunctional activation, termed immunosenescence, compared with those from younger hosts, and this may be in part due to mitochondrial dysfunction that increases with age [Citation112]. However, in cancer types that respond more robustly to T cell checkpoint inhibitors, the effect of age on outcomes is not clear – in non-small cell lung cancer, advanced age was associated with worse responses to these agents [Citation113], while in melanoma, age may not affect outcomes [Citation114]. Whether patient age will become an important variable in prostate cancer immunity is uncertain but should be considered.
Beyond aging, lymphocytes from patients with prostate cancer are also subjected to the effects of androgen deprivation. Surgical or chemical castration leads to thymic enlargement due to the loss of negative regulation of thymic epithelial cells [Citation115,Citation116] and increased peripheral naïve T cells that exhibit enhanced activation [Citation117]. In prostate cancer, castration leads to expansion of tumor-specific T cells in the Pb-HAx TRAMP model [Citation118] and increased density of TILs in patients [Citation119]. Although men generally experience less autoimmunity than women, the cumulative effect of testosterone on tumor immunity is not clear – while testosterone was shown to inhibit differentiation of CD4 + T cells to the pro-inflammatory Th1 subtype via upregulation of Ptpn1 and increase production of the immunosuppressive cytokine IL-10 [Citation120,Citation121], it was also found to stimulate antitumor neutrophil function in melanoma mouse models [Citation122]. In a recent clinical trial of sipuleucel-T given either in the castrate or noncastrate setting in men with biochemically recurrent prostate cancer, antitumor antigen-specific immune responses were greater when the immunotherapy was used in the noncastrate setting [Citation123]. The effect of testosterone on tumor immunity is a critical area for future investigation. Another point to consider is that the immune effects of castration are likely not equivalent to those of antiandrogens, such as enzalutamide. A well-described side-effect of enzalutamide is seizure, due to off-target inhibition of γ-aminobutyric acid type A (GABA-A) receptors in the brain. Interestingly, enzalutamide was also shown to inhibit T cell activation via inhibition of the GABA-A receptor [Citation124]. Ongoing trials testing the combination of enzalutamide with ICI (e.g., NCT03834493, NCT04191096) will elucidate whether a direct effect of enzalutamide on tumor will offset a possible negative effect on T cell activation to allow for efficacy of this drug combination.
Alternate T cell checkpoints in prostate cancer. Regulation of T cell activation is so critical to preserve self-tolerance and avoid autoimmunity that there are multiple layers of redundancy. As mentioned, CTLA4 and PD1 are only two of several T cell checkpoints. Two additional checkpoints that may be functionally important in prostate cancer are the A2A adenosine receptor, EZH2 (Enhancer of zeste homolog 2), and the so-far undefined receptor of B7-H3 [Citation125]. It is theoretically possible that these checkpoints eclipse roles for CTLA4 and PD1 in prostate cancer immunity.
Damaged or dying tumor cells release ATP, which may function as a DAMP. Over time, ATP is converted to AMP and adenosine by ectonucleotidases, such as CD73, which subsequently suppress immune responses by binding the adenosine A2A receptor expressed on T cells and NK cells, and inhibiting effector function [Citation126]. High CD73 expression in normal prostate epithelium adjacent to tumor correlated with shorter biochemical recurrence in patients who had prostatectomy [Citation127], and CD73 expression in prostate tumor cells was enhanced in those known to have metastasized to lymph nodes [Citation128]. Additionally, PAP can function as an ectonucleotidase to contribute to pools of adenosine in the prostate tumor microenvironment [Citation129]. Multiple trials of A2A receptor inhibitors in combination with PD1 inhibitors and/or CD73 inhibitors are underway and listed in .
EZH2 is an epigenetic regulator that was found to have increased expression in T cells of patients treated with and resistant to ipilimumab [Citation130]. Inhibition of EZH2 increased prostate cancer tumor immunity and synergized with ipilimumab in murine models. Beyond EZH2 function in T cells, a recent study further suggests that EZH2 inhibition may induce dsRNA intracellular stress and increased STING-ISG response within prostate cancer cells, altering the tumor immune microenvironment, and augmenting cancer cell T cell killing [Citation131]. The combination of the EZH1/2 inhibitor DS3201 and nivolumab is being testing in phase 1 clinical trials (NCT04388852).
Among the B7 superfamily, B7-H3 (less commonly known as PD-L3) appears to be more highly expressed than PD-L1, PD-L2, and B7-H4 in prostate cancer, and its expression correlates with Gleason score, tumor stage, and castration-resistant metastatic disease, [Citation53,Citation125]. In a recent study of metastatic prostate cancer, up to 88% of the samples had a high expression of B7-H3, with expression correlating with AR expression [Citation55]. The function of B7-H3 is poorly defined; it is unknown whether it can activate co-stimulatory pathways or co-inhibitory pathways, or perhaps this may be context-dependent. Interestingly, unlike the other checkpoints PD-L1, PD-L2, and the adenosine A2A receptor, its expression was found to be negatively associated with the dMMR mutational signature but positively associated with the HRR mutational signature [Citation53]. Clinical trials are ongoing with enoblituzumab (formerly MGA271), a humanized anti-B7-H3 monoclonal antibody, for patients with advanced cancer that expresses B7-H3 [Citation132] and for patients with prostate cancer specifically (NCT02923180).
Prostate cancer microenvironment is immunosuppressive. Although the CA184-043 study suggested that patients with lower tumor burden may obtain greater benefit from immune checkpoint blockade, this has not been rigorously assessed in subsequent trials of prostate cancer. In melanoma and non-small cell lung cancer, a lower baseline tumor size is associated with improved response rates and overall survival following treatment with ICI monotherapy [Citation133,Citation134]. Indeed, in both of these tumor types, T cell checkpoint inhibitor monotherapy provides a significant OS benefit when administered following local therapy when the tumor burden is lowest [Citation135,Citation136]. Mechanistically, this may support the idea that the tumor microenvironment associated with bulky tumors is immunosuppressive. This immunosuppression may be driven by tumor depletion of factors required for effective immune cell function, such as oxygen and metabolites, or by tumor production of factors that directly suppress immune cell function, such as TGF-β, adenosine, or kynurenine.
Prostate cancer appears to have a particularly immunosuppressive microenvironment [Citation137]. It is known to have a relatively high number of infiltrating CD4+ and CD8+ regulatory T cells (Tregs) compared with other cancer types [Citation138], and secreted factors appear to induce differentiation of TILs into these suppressive subtypes [Citation138–140]. Androgen deprivation induces T cell infiltration in prostate cancers [Citation141]; however, many of these immune cells are found to be Tregs [Citation142,Citation143]. Thus, strategies to impair or deplete Tregs beyond CTLA4 and PD1 inhibition should be considered. For example, CDK4/6 inhibitors have been shown to both induce breast cancer cell cycle arrest as well as stimulate an antitumor immune response [Citation144]. Interestingly, CDK4/6 inhibitors both stimulated tumor-cell IFNɣ signaling and antigen presentation as well-inhibited Treg proliferation. The combination of abemaciclib and atezolizumab is currently being tested in a phase 2 clinical trial for mCRPC (NCT04751929).
It remains to be determined precisely how prostate cancer induces differentiation of effector T cells into suppressive subtypes. Increasing evidence suggests that tumor cell metabolism not only supports the energetic and biosynthetic needs of the tumor but also modifies intra- and inter-cellular signaling pathways to mediate immune evasion [Citation145]. Prostate cancer seems to engage in higher rates of fatty acid synthesis compared with other cancer types and normal prostate, which appears to be driven by AR and c-Myc [Citation146–150]. In contrast, immunosuppressive cell subtypes, including Tregs and MDSCs, are known to oxidize fatty acids [Citation151–153]. Tregs express high levels of CD36 that allow for uptake of fatty acids from the environment, which is required for their suppressive function [Citation154]. Thus, a possibility is that production of fatty acids is a mechanism of immune suppression in prostate cancer. The PPARα inhibitor TPST-1120 is currently in phase I clinical trials in combination with nivolumab for advanced solid tumors (NCT03829436). This agent is purported to reduce fatty acid oxidation in immune cells, decreasing differentiation to immunosuppressive subtypes. Interestingly, androgens were described to stimulate PPARα expression in lymphocytes, reducing autoimmunity in male compared to female mice [Citation155].
Prostate cancer insensitivity to immunogenic cell death. Finally, in order to be visible to CD8+ cytotoxic T cells, neoantigens must be presented on the tumor cell surface by major histocompatibility complex (MHC) proteins. However, studies dating back several decades indicate that prostate cancer may demonstrate low expression of MHC class I as 30–50% of the primary prostate cancers and up to 80% of metastatic prostate cancers were shown to exhibit complete loss of MHC class I [Citation156–158]. Interestingly, up to 60% of primary prostate cancer may express MHC class II [Citation159], which is recognized by CD4 + T cells. While MHC class II is traditionally associated with expression on professional APCs, there is a growing appreciation that it can also be expressed by tumor cells and play an important role in tumor immunity [Citation160]; however, how to harness this molecule for therapeutic benefit is not clear. MHC expression can be induced in tumor cells through IFNγ signaling, perhaps as by CDK4/6 inhibitors. In prostate cancer models, BET/bromodomain inhibition was shown to increase expression of MHC class I and was suggested to augment ICB efficacy [Citation161]. Additionally, BET inhibition may improve T memory cell formation [Citation162]. The BET inhibitor ZEN-3694 is currently being tested in a phase 2 clinical trial for mCRPC in combination with enzalutamide and pembrolizumab (NCT04471974).
12. Conclusion
While CTLA4 and PD1 inhibition may be effective for a small molecular subset of patients with prostate cancer [Citation163], it seems that the majority of prostate cancers possess barriers to an adaptive immune response that are not overcome by these agents. These barriers may constitute alternate immune checkpoints that possess greater significance for this disease. Moreover, these barriers may occur early in the tumor-immunity cycle, such as in the production or recognition of tumor antigen and the activation of an innate immune response. Additionally, there may be T cell checkpoints beyond CTLA4 and PD1, including alternate co-inhibitory receptors and hormonal and metabolic factors that are activated and may be important in prostate cancer. Given the likelihood that multiple immune checkpoints are activated by prostate cancer, future immunotherapeutic strategies should aim to target the spectrum of immune checkpoints in this disease, which is most likely to be achieved by combination therapies. Although we highlighted targeting alternate immune checkpoints in combination with CTLA4 and PD1 blockade, many rational permutations and layering of combination therapies should be considered.
Article highlights
CTLA4 and PD1 blockade can stimulate antitumor immune responses leading to tumor regression in several cancer types, but have limited efficacy in unselected patients with prostate cancer.
Activity of these agents may be limited by a poor endogenous adaptive immune response to prostate cancer.
Barriers to an adaptive immune response include low tumoral production of neoantigens, impaired innate immune cell function and T cell priming, and impaired T cell function.
We review how these barriers could be overcome by combination therapies to stimulate the elusive durable immune response to advanced prostate cancer.
Declaration of interest
ESA has served as a paid consultant/advisor for Janssen, Pfizer, Sanofi, Dendreon, Merck, Bristol–Myers Squibb, AstraZeneca, Clovis, Eli Lilly and Amgen; has received research funding to his institution from Janssen, Johnson & Johnson, Sanofi, Dendreon, Genentech, Novartis, Merck, Bristol–Myers Squibb, AstraZeneca and Constellation; and is a co-inventor of an AR-V7 biomarker technology that has been licensed to Qiagen. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The authors apologize to those investigators whose important work we did not discuss for the sake of conciseness.
Additional information
Funding
References
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264.
- Schildberg FA, Klein SR, Freeman GJ, et al. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity. 2016;44(5):955–972.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736.
- Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–988.
- Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–547.
- Lo B, Abdel-Motal UM. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr Opin Immunol. 2017;49:14–19.
- Ise W, Kohyama M, Nutsch KM, et al. CTLA-4 suppresses the pathogenicity of self antigen-specific T cells by cell-intrinsic and cell-extrinsic mechanisms. Nat. Immunol. 2010;11(2):129–135.
- Perkins D, Wang Z, Donovan C, et al. Regulation of CTLA-4 expression during T cell activation. J. Immunol. 1996;156(11):4154–4159.
- Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009;229(1):12–26.
- Tai X, Van Laethem F, Sharpe AH, et al. Induction of autoimmune disease in CTLA-4-/- mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc. Natl. Acad. Sci. U. S. A. 2007;104(34):13756–13761.
- Kong K-F, Fu G, Zhang Y, et al. Protein kinase C-η controls CTLA-4-mediated regulatory T cell function. Nat. Immunol. 2014;15(5):465–472.
- Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–275 .
- Peggs KS, Quezada SA, Chambers CA, et al. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009;206(8):1717–1725.
- van Elsas A, Sutmuller RP, Hurwitz AA, et al. Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: comparison of prophylaxis and therapy. J. Exp. Med. 2001;194(4):481–489.
- Fassò M, Waitz R, Hou Y, et al. SPAS-1 (stimulator of prostatic adenocarcinoma-specific T cells)/SH3GLB2: a prostate tumor antigen identified by CTLA-4 blockade. Proc. Natl. Acad. Sci. U. S. A. 2008;105(9):3509–3514.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–581 .
- Wei SC, Levine JH, Cogdill AP, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170(6):1120–1133.e17.
- Simpson TR, Li F, Montalvo-Ortiz W, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013;210(9):1695–1710.
- Pai -C-CS, Simons DM, Lu X, et al. Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J. Clin. Invest. 2019;129(1):349–363.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
- Keir ME, Liang SC, Guleria I, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006;203(4):883–895.
- Chemnitz JM, Parry RV, Nichols KE, et al. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004;173(2):945–954.
- Sheppard K-A, Fitz LJ, Lee JM, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574(1–3):37–41.
- Wei F,Zhong S, Ma Z, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc. Natl. Acad. Sci. U. S. A. 2013;110(27):E2480–9.
- Chang C-H, Curtis JD, Maggi LB Jr, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239–1251.
- Patsoukis N, Bardhan K, Chatterjee P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015;6(1):6692.
- Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009;206(13):3015–3029.
- Strauss L, Mahmoud MAA, Weaver JD, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020;5(43):eaay1863.
- Nishimura H,Okazaki T, Tanaka Y, et al.Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291(5502):319–322.
- Nishimura H, Nose M, Hiai H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–151.
- Wang J, Yoshida T, Nakaki F, et al. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. U. S. A. 2005;102(33):11823–11828.
- Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016;375(9):819–829.
- Gao J, Shi LZ, Zhao H, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404.e9.
- Manguso RT, Pope HW, Zimmer MD, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547(7664):413–418.
- Kwon ED, Drake CG, Scher HI, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700–712.
- Fizazi K, Drake CG, Beer TM, et al.Final analysis of the ipilimumab versus placebo following radiotherapy phase III trial in postdocetaxel metastatic castration-resistant prostate cancer identifies an excess of long-term survivors. Eur. Urol. 2020;78(6):822–830.
- Beer TM, Kwon ED, Drake CG, et al. randomized, double-blind, phase iii trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J. Clin. Oncol. 2017;35(1):40–47.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012;366(26):2443–2454.
- Graff JN, Alumkal JJ, Drake CG, et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget. 2016;7(33):52810–52817.
- Hansen AR,Massard C, Ott PA, et al. Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann. Oncol. 2018;29(8):1807–1813.
- Antonarakis ES, Piulats JM, Gross-Goupil M, et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020;38(5):395–405.
- Hoimes CJ, Graff JN, Tagawa ST, et al. KEYNOTE-199 cohorts (C) 4 and 5: phase II study of pembrolizumab (pembro) plus enzalutamide (enza) for enza-resistant metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020;38(15_suppl):5543.
- Boudadi K, Suzman DL, Anagnostou V, et al. Ipilimumab plus nivolumab and DNA-repair defects in AR-V7-expressing metastatic prostate cancer. Oncotarget. 2018;9(47):28561–28571.
- Sharma P,Pachynski RK, Narayan V, et al. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the CheckMate 650 trial. Cancer Cell. 2020;38(4):489–499.e3.
- Gao J, Ward JF, Pettaway CA, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017;23(5):551–555.
- Trujillo JA, Sweis RF, Bao R,Sweis RF, Bao R, et al. T cell-inflamed versus non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol. Res. 2018;6(9):990–1000.
- Ji -R-R, Chasalow SD, Wang L, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012;61(7):1019–1031.
- Subudhi SK, Vence L, Zhao H, et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci. Transl. Med. 2020;12(537):eaaz3577.
- Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127(8):2930–2940.
- de Bono JS, Guo C, Gurel B, et al. Prostate carcinogenesis: inflammatory storms. Nat Rev Cancer. 2020;20(8):455–469.
- Tamborero D, Rubio-Perez C, Muiños F, et al. A pan-cancer landscape of interactions between solid tumors and infiltrating immune cell populations. Clin. Cancer Res. 2018;24(15):3717–3728.
- Rodrigues DN, Rescigno P, Liu D, et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. Journal of Clinical Investigation. 2018;128(10):4441–4453.
- Mendonca J, Kumar R, Owoyemi O, et al. Supraphysiological testosterone induces ferroptosis and activates NF-kappaB mediated immune pathways in prostate cancer through nucleophagy. bioRxiv. 2020; DOI:https://doi.org/10.1101/2020.09.10.286252.
- Brady L, Kriner M, Coleman I, et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat. Commun. 2021;12(1). DOI:https://doi.org/10.1038/s41467-021-21615-4.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571 .
- He MX, Cuoco MS, Crowdis J, et al. Transcriptional mediators of treatment resistance in lethal prostate cancer. bioRxiv. 2020; DOI:https://doi.org/10.1101/2020.03.19.998450.
- Samstein RM, Lee C-H, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019;51(2):202–206.
- Li G-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98.
- Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413.
- Mehnert JM, Panda A, Zhong H, et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J. Clin. Invest. 2016;126(6):2334–2340.
- Gong J, Wang C, Lee PP, et al. Response to PD-1 Blockade in Microsatellite Stable Metastatic Colorectal Cancer Harboring a POLE Mutation. J. Natl. Compr. Canc. Netw. 2017;15(2):142–147.
- Lee L, Ali S, Genega E, et al. Aggressive-variant microsatellite-stable POLE mutant prostate cancer with high mutation burden and durable response to immune checkpoint inhibitor therapy. JCO Precis Oncol. 2018;1–8.
- Ryan MJ, Bose R. Genomic alteration burden in advanced prostate cancer and therapeutic implications. Front Oncol. 2019;9(1287). DOI:https://doi.org/10.3389/fonc.2019.01287
- Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218.
- Sena LA, Fountain J, Isaacsson Velho P, et al. Tumor frameshift mutation proportion predicts response to immunotherapy in mismatch repair-deficient prostate cancer. Oncologist. 2021;26(2):e270–e278; DOI:https://doi.org/10.1002/onco.13601.
- Graham LS, Montgomery B, Cheng HH, et al. Mismatch repair deficiency in metastatic prostate cancer: response to PD-1 blockade and standard therapies. PLOS ONE. 2020;15(5):e0233260.
- Barata P, Agarwal N, Nussenzveig R, et al. Clinical activity of pembrolizumab in metastatic prostate cancer with microsatellite instability high (MSI-H) detected by circulating tumor DNA. J Immunother Cancer. 2020;8(2):e001065.
- Abida W, Cheng ML, Armenia J, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 2019;5(4):471–478.
- Antonarakis ES, Shaukat F, Isaacsson Velho P, et al. Clinical Features and Therapeutic Outcomes in Men with Advanced Prostate Cancer and DNA Mismatch Repair Gene Mutations. Eur. Urol. 2019;75(3):378–382.
- Wu Y-M, Cieślik M, Lonigro RJ, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. 2018;173(7):1770–1782.e14.
- Antonarakis ES. Cyclin-dependent kinase 12, immunity, and prostate cancer. N. Engl. J. Med. 2018;379(11):1087–1089.
- Antonarakis ES, Isaacsson Velho P, Fu W, et al. CDK12 -Altered Prostate Cancer: clinical Features and Therapeutic Outcomes to Standard Systemic Therapies, Poly (ADP-Ribose) Polymerase Inhibitors, and PD-1 Inhibitors . JCO Precis Oncol. 2020;4(4):370–381 .
- Schweizer MT, Ha G, Gulati R, et al. CDK12-Mutated Prostate Cancer: clinical Outcomes With Standard Therapies and Immune Checkpoint Blockade. JCO Precis Oncol. 2020;4(4):382–392.
- Samstein RM, Krishna C, Ma X, et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat Cancer. 2020;1(12):1188–1203.
- Linnebacher M, Gebert J, Rudy W, et al. Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int J Cancer. 2001;93(1):6–11.
- Turajlic S, Litchfield K, Xu H, et a. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18(8):1009–1021.
- O’Donnell T, Christie EL, Ahuja A, et al. Chemotherapy weakly contributes to predicted neoantigen expression in ovarian cancer. BMC Cancer. 2018;18(1):87.
- Sheikh NA, Petrylak D, Kantoff PW, et al. Sipuleucel-T immune parameters correlate with survival: an analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol. Immunother. 2013;62(1):137–147.
- Fong L, Carroll P, Weinberg V, et al. Activated lymphocyte recruitment into the tumor microenvironment following preoperative sipuleucel-T for localized prostate cancer. J. Natl. Cancer Inst. 2014;106(11). DOI:https://doi.org/10.1093/jnci/dju268.
- Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010;363(5):411–422.
- Scholz M, Yep S, Chancey M, et al. Phase I clinical trial of sipuleucel-T combined with escalating doses of ipilimumab in progressive metastatic castrate-resistant prostate cancer. ImmunoTargets Ther. 2017;6:11–16.
- McNeel DG, Eickhoff JC, Wargowski E, et al. Concurrent, but not sequential, PD-1 blockade with a DNA vaccine elicits anti-tumor responses in patients with metastatic, castration-resistant prostate cancer. Oncotarget. 2018;9(39):25586–25596.
- McNeel DG, Becker JT, Eickhoff JC, et al. Real-time immune monitoring to guide plasmid DNA vaccination schedule targeting prostatic acid phosphatase in patients with castration-resistant prostate cancer. Clin. Cancer Res. 2014;20(14):3692–3704.
- Kyriakopoulos CE, Eickhoff JC, Ferrari AC, et al. Multicenter phase I trial of a DNA vaccine encoding the androgen receptor ligand-binding domain (pTVG-AR, MVI-118) in patients with metastatic prostate cancer. Clin Cancer Res off J Am Assoc Cancer Res. 2020;26(19):5162–5171.
- Hildner K, Edelson BT, Purtha WE, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–1100.
- Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med. 2011;208(10):2005–2016.
- Woo S-R, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015;33(1):445–474.
- Guerra N, Tan YX, Joncker NT, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28(4):571–580.
- Chin AI, Miyahira AK, Covarrubias A, et al. Toll-like receptor 3-mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res. 2010;70(7):2595–2603.
- van Soest RJ, Templeton AJ, Vera-Badillo FE, et al. Neutrophil-to-lymphocyte ratio as a prognostic biomarker for men with metastatic castration-resistant prostate cancer receiving first-line chemotherapy: data from two randomized phase III trials. Ann. Oncol. 2015;26(4):743–749.
- Conteduca V, Crabb SJ, Jones RJ, et al. Persistent Neutrophil to Lymphocyte Ratio >3 during Treatment with Enzalutamide and Clinical Outcome in Patients with Castration-Resistant Prostate Cancer. PLoS One. 2016;11(7):e0158952.
- Zarif JC, Baena-Del Valle JA, Hicks JL, et al. Mannose receptor-positive macrophage infiltration correlates with prostate cancer onset and metastatic castration-resistant disease. Eur. Urol. Oncol. 2019;2(4):429–436.
- Lundholm M, Hägglöf C, Wikberg ML, et al. Secreted factors from colorectal and prostate cancer cells skew the immune response in opposite directions. Sci. Rep. 2015;5(1):15651.
- Di Mitri D, Mirenda M, Vasilevska J, et al. Re-education of tumor-associated macrophages by CXCR2 blockade drives senescence and tumor inhibition in advanced prostate cancer. Cell Rep. 2019;28(8):2156–2168.e5.
- Jaynes JM, Sable R, Ronzetti M, et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 2020;12(530):eaax6337.
- Kodumudi KN, Woan K, Gilvary DL, et al. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin. Cancer Res. 2010;16(18):4583–4594.
- Lu X, Horner JW, Paul E, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543(7647):728–732.
- Patnaik A, Swanson KD, Csizmadia E, et al. Cabozantinib eradicates advanced Murine prostate cancer by activating antitumor innate immunity. Cancer Discov. 2017;7(7):750–765.
- Agarwal N, Loriot Y, McGregor BA, et al. Cabozantinib (C) in combination with atezolizumab (A) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): results of Cohort 6 of the COSMIC-021 Study. J. Clin. Oncol. 2020;38(6_suppl):139.
- Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548.
- Jairath NK, Farha MW, Srinivasan S, et al. Tumor immune microenvironment clusters in localized prostate adenocarcinoma: prognostic impact of macrophage enriched/plasma cell non-enriched subtypes. J. Clin. Med. 2020;9(6):1973.
- Weiner AB, Vidotto T, Liu Y, et al. Plasma cells are enriched in localized prostate cancer in Black men and are associated with improved outcomes. Nat. Commun. 2021;12(1). DOI:https://doi.org/10.1038/s41467-021-21245-w.
- Gajewski TF, Higgs EF. Immunotherapy with a sting. Science. 2020;369(6506):921–922.
- Woo S-R, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2015;42(1):199 .
- Ager CR, Reilley MJ, Nicholas C, et al. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol. Res. 2017;5(8):676–684.
- Ding L, Kim H-J, Wang Q, et al. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 2018;25(11):2972–2980.e5.
- Reisländer T, Lombardi EP, Groelly FJ, et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019;10(1). DOI:https://doi.org/10.1038/s41467-019-11048-5.
- Shen J, Zhao W, Ju Z, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79(2):311–319.
- Denmeade SR, Wang H, Agarwal N, et al. TRANSFORMER: a randomized phase II study comparing bipolar androgen therapy versus enzalutamide in asymptomatic men with castration-resistant metastatic prostate cancer. J Clin Oncol. 2021;JCO2002759.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363(8):711–723.
- Desdín-Micó G, Soto-Heredero G, Aranda JF, et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science. 2020;368(6497):1371–1376.
- Lichtenstein MRL, Nipp RD, Muzikansky A, et al. Impact of age on outcomes with immunotherapy in patients with non-small cell lung cancer. J. Thorac. Oncol. 2019;14(3):547–552.
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22(8):963–971.
- Heng TSP, Goldberg GL, Gray DHD, et al. Effects of castration on thymocyte development in two different models of thymic involution. J. Immunol. 2005;175(5):2982–2993.
- Sutherland JS, Goldberg GL, Hammett MV, et al. Activation of thymic regeneration in mice and humans following androgen blockade. J. Immunol. 2005;175(4):2741–2753.
- Viselli SM, Stanziale S, Shults K, et al. Castration alters peripheral immune function in normal male mice. Immunology. 1995;84(2):337–342.
- Drake CG, Doody ADH, Mihalyo MA, et al Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer Cell. 2005;7(3):239–249.
- Obradovic AZ, Dallos MC, Zahurak ML, et al. T-Cell Infiltration and Adaptive Treg Resistance in Response to Androgen Deprivation With or Without Vaccination in Localized Prostate Cancer. Clin. Cancer Res. 2020;26(13):3182–3192.
- Kissick HT, Sanda MG, Dunn LK, et al. Androgens alter T-cell immunity by inhibiting T-helper 1 differentiation. Proc. Natl. Acad. Sci. U. S. A. 2014;111(27):9887–9892.
- Liva SM, Voskuhl RR. Testosterone Acts Directly on CD4+ T Lymphocytes to Increase IL-10 Production. J. Immunol. 2001;167(4):2060–2067.
- Markman JL, Porritt RA, Wakita D, et al. Loss of testosterone impairs anti-tumor neutrophil function. Nat. Commun. 2020;11(1). DOI:https://doi.org/10.1038/s41467-020-15397-4.
- Antonarakis ES, Kibel AS, Yu EY, et al. Sequencing of sipuleucel-T and androgen deprivation therapy in men with hormone-sensitive biochemically recurrent prostate cancer: a phase II randomized trial. Clin. Cancer Res. 2017;23(10):2451–2459.
- Pu Y, Xu M, Liang Y, et al. Androgen receptor antagonists compromise T cell response against prostate cancer leading to early tumor relapse. Sci. Transl. Med. 2016;8(333):333ra47–333ra47.
- Benzon B, Zhao SG, Haffner MC, et al. Correlation of B7-H3 with androgen receptor, immune pathways and poor outcome in prostate cancer: an expression-based analysis. Prostate Cancer Prostatic Dis. 2017;20(1):28–35.
- Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. 2018;6(1). https://doi.org/10.1186/s40425-018-0360-8
- Leclerc BG, Charlebois R, Chouinard G, et al. CD73 expression is an independent prognostic factor in prostate cancer. Clin. Cancer Res. 2016;22(1):158–166.
- Yang Q, Du J, Zu L. Overexpression of CD73 in prostate cancer is associated with lymph node metastasis. Pathol. Oncol. Res. 2013;19(4):811–814.
- Zylka MJ, Sowa NA, Taylor-Blake B, et al. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron. 2008;60(1):111–122.
- Goswami S, Apostolou I, Zhang J, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Invest. 2018;128(9):3813–3818.
- Morel KL, Sheahan AV, Burkhart DL, et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer. 2021;2(4):444–456.
- Powderly J, Cote G, Flaherty K, et al. Interim results of an ongoing Phase I, dose escalation study of MGA271 (Fc-optimized humanized anti-B7-H3 monoclonal antibody) in patients with refractory B7-H3-expressing neoplasms or neoplasms whose vasculature expresses B7-H3. J Immunother Cancer. 2015;3(Suppl 2):O8.
- Joseph RW, Elassaiss-Schaap J, Kefford R, et al. Baseline tumor size is an independent prognostic factor for overall survival in patients with melanoma treated with pembrolizumab. Clin. Cancer Res. 2018;24(20):4960–4967.
- Katsurada M, Nagano T, Tachihara M, et al. Baseline tumor size as a predictive and prognostic factor of immune checkpoint inhibitor therapy for non-small cell lung cancer. Anticancer Res. 2019;39(2):815–825.
- Eggermont AMM, Chiarion-Sileni V, Grob J-J, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N. Engl. J. Med. 2016;375(19):1845–1855.
- Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N. Engl. J. Med. 2017;377(20):1919–1929.
- Vitkin N, Nersesian S, Siemens DR, et al. The tumor immune contexture of prostate cancer. Front Immunol. 2019;10(603). https://doi.org/10.3389/fimmu.2019.00603
- Kiniwa Y, Miyahara Y, Wang HY, et al. CD8 + Foxp3+Regulatory T Cells Mediate Immunosuppression in Prostate Cancer. Clin. Cancer Res. 2007;13(23):6947–6958.
- Getnet D, Maris CH, Hipkiss EL, et al. Tumor recognition and self-recognition induce distinct transcriptional profiles in antigen-specific CD4 T cells. J. Immunol. 2009;182(8):4675–4685.
- Shafer-Weaver KA, Anderson MJ, Stagliano K, et al. Cutting Edge: tumor-Specific CD8 + T Cells Infiltrating Prostatic Tumors Are Induced to Become Suppressor Cells. J. Immunol. 2009;183(8):4848–4852.
- Mercader M, Bodner BK, Moser MT, et al. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 2001;98(25):14565–14570.
- Sorrentino C, Musiani P, Pompa P, et al. Androgen deprivation boosts prostatic infiltration of cytotoxic and regulatory T lymphocytes and has no effect on disease-free survival in prostate cancer patients. Clin. Cancer Res. 2011;17(6):1571–1581.
- Tang S, Moore ML, Grayson JM, et al. Increased CD8 + T-cell Function following Castration and Immunization Is Countered by Parallel Expansion of Regulatory T Cells. Cancer Res. 2012;72(8):1975–1985.
- Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–475.
- Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer. 2020;20(9):516–531.
- Reznik E, Luna A, Aksoy BA, et al. A landscape of metabolic variation across tumor types. Cell Syst. 2018;6(3):301–313.e3.
- Massie CE, Lynch A, Ramos-Montoya A, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30(13):2719–2733.
- Han W, Gao S, Barrett D, et al. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene. 2018;37(6):710–721.
- Singh KB, Hahm E-R, Singh SV. Abstract 831: c-Myc is a novel target of prostate cancer cell growth inhibition by honokiol. Cancer Res. 2016;76(4):831.
- Sena LA, Denmeade SR. Fatty acid synthesis in prostate cancer: vulnerability or epiphenomenon? Cancer Res. 2021;canres.1392.2021.
- Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting Edge: distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4 +T Cell Subsets. J. Immunol. 2011;186(6):3299–3303 .
- Gerriets VA, Kishton RJ, Nichols AG, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest. 2015;125(1):194–207.
- Hossain F, Al-Khami AA, Wyczechowska D, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 2015;3(11):1236–1247.
- Wang H, Franco F, Tsui Y-C, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020;21(3):298–308.
- Dunn SE, Ousman SS, Sobel RA, et al. Peroxisome proliferator–activated receptor (PPAR)α expression in T cells mediates gender differences in development of T cell–mediated autoimmunity. J. Exp. Med. 2007;204(3):693.
- Blades RA, Keating PJ, McWilliam LJ, et al. Loss of HLA class I expression in prostate cancer: implications for immunotherapy. Urology. 1995;46(5):681–686. discussion 686-7.
- Bander NH, Yao D, Liu H, et al. MHC class I and II expression in prostate carcinoma and modulation by interferon-alpha and -gamma. Prostate. 1997;33(4):233–239.
- Carretero FJ, Del Campo AB, Flores-Martín JF, et al. Frequent HLA class I alterations in human prostate cancer: molecular mechanisms and clinical relevance. Cancer Immunol. Immunother. 2016;65(1):47–59.
- Younger AR, Amria S, Jeffrey WA, et al. HLA class II antigen presentation by prostate cancer cells. Prostate Cancer Prostatic Dis. 2008;11(4):334–341.
- Axelrod ML, Cook RS, Johnson DB, et al. Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 2019;25(8):2392–2402.
- Mao W, Ghasemzadeh A, Freeman ZT, et al. Immunogenicity of prostate cancer is augmented by BET bromodomain inhibition. J Immunother Cancer. 2019;7(1). https://doi.org/10.1186/s40425-019-0758-y.
- Kagoya Y, Nakatsugawa M, Yamashita Y, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Invest. 2016;126(9):3479–3494.
- Antonarakis ES. A new molecular taxonomy to predict immune checkpoint inhibitor sensitivity in prostate cancer. Oncologist. 2019;24(4):430–432.