Abstract
Utilizing green technologies as microwave (MW) irradiation and ionic liquids (ILs), we could produce 1,3,5-trisubstituted benzene 3, 7, 9, and 14 by self-condensation of enamines 1, enaminones 4, 8, and enaminoester 10, respectively, in the presence of pyridinium chloride ([PyH]Cl) for short time. Also, we synthesized pyridine derivatives 17, upon irradiating enaminones 4 in domestic MW oven for short time.
Introduction
Adopting green methodologies for synthesis of polyfunctional aromatics and heteroaromatics is now receiving considerable attention Citation1–3. In the last decade, we were interested in exploring potentialities of enamines and we could already discover several novel synthesis of polyfunctional aromatics and heteroaromatics utilizing enamines as starting materials Citation4–6. Recently, we have been interested in adopting green methodologies to our synthetic approaches and we could efficiently adopt MW irradiation (MW) as energy source for quite a number of our synthesis Citation3 Citation7–11. In the present article, we report the utility of domestic MW as energy source and ionic liquid (IL) as solvent for self-condensation of enamines, reported recently from our laboratories via refluxing the enamines in acetic acid, in presence or absence of ammonium ion for several hours Citation12–14. We observed that with this technique, high boiling solvents evaporate producing some hazard. To avoid this complication we thought to adopt microwave heating in IL trying to conduct the reactions of enamines and enaminones in a green way. ILs are one of several technologies used in green chemistry that aimed to reduce environmental harmful effect of chemicals like organovolatile solvent Citation15. Many of these solvents are known to upset our ecosystems by depleting the ozone layer and participating in the reactions that form tropospheric smog. In addition, some solvents are neurotoxins, may cause sterility, or may cause cancer. While continuous using of these solvents would not be acceptable from both an environmental and a health perspective, such operations are difficult to achieve, and alternative solvents are currently being sought to minimize the problems inherent in solvent release to the environment. ILs are believed to have minimum vapor pressure and have been observed to increase MW energy absorption Citation16. Therefore, several MW reactions have been already successfully conducted in ILs Citation17.
Results and discussion
The substituted enamines 1a–f, enaminones 4a–j, 8, and enaminoesters 10a–c were prepared and their structures were further investigated in the light of reported existence of 8 in Z-form Citation18. Contradicting all previous reports on structure of enaminones Citation19–21, now we confirm that this enaminone exists in trans form as indicated earlier by Al-Mousawi Citation22.
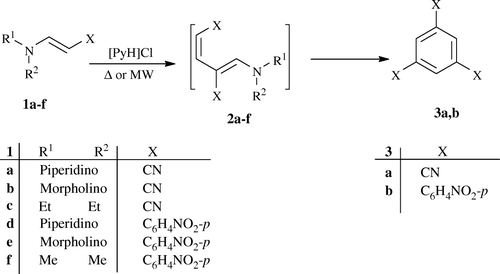
Previously, it has been reported that enamines 1a–c gave only the open chain compounds 2a–c upon treatment with AcOH Citation23. In our hands, heating enamines 1a–f in [PyH]Cl afforded 1,3,5-trisubstituted benzene 3a,b as the only reaction products ().
Enaminones 4a–j, prepared by reacting methylketones with dimethyl formamid dimethyl acetal (DMFDMA) in MW oven Citation24, underwent self-condensation on heating at 110°C in [PyH]Cl, as an IL, for 20 min or irradiation at 400 watt, 105°C for 1 min in a modified domestic MW oven yielding 1,3,5-trisubstituted benzene 7a–j () in high yield ().
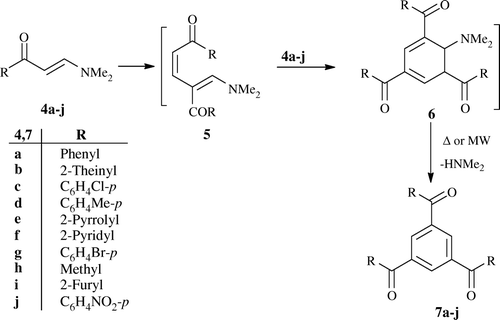
Table 1. Comparing yield of 7a–j using thermal and microwave irradiation in the presence of IL.
From one can see that there is an improvement in rates and yields of reactions under both conventional heating and MW in IL, compared with classical condition in reported literature Citation12.
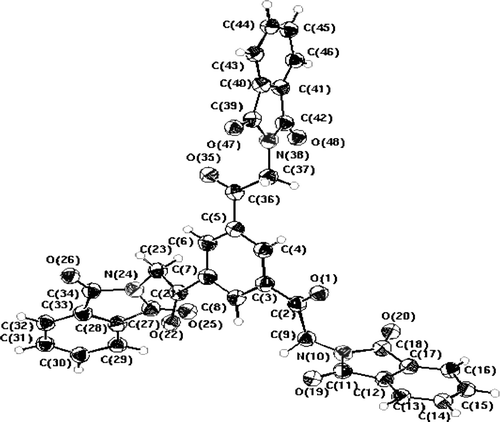
We believe that 4a–j is initially converted to the open chain intermediate 5 that then adds further one molecule of 4a–j to yield the intermediate 6 that is aromatized, under these reaction conditions, to the final isolable products 7a–j. Similarly, compound 8 afforded 9 under the same reaction conditions (). The structure of 9 was established for the reaction product on the basis of its elemental analysis and spectral data (MS, IR, 1H NMR; see Section “Experimental”). Finally, the structure of 9 was unambiguously confirmed by X-ray crystallography ().Footnote1
Figure 1. Molecular structure of 9 with atoms labeling scheme.
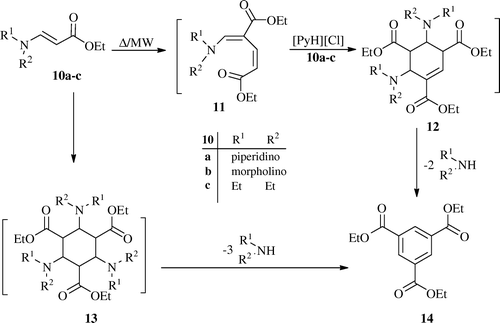
In the same way, enaminoesters 10a–c underwent self-condensation to yield triethylbenzene-1,3,5-tricarboxylat 14. The formation of 14 is assumed to proceed via self-condensation of 10 to yield the intermediate 11 which reacts with another molecule of 10 to give another intermediate 12 which then loses two molecules of secondary amines (piperidine, morpholine, or diethylamine) to give the final product 14 (). Alternatively, compound 14 may be formed through [2+2+2] cycloaddition of 10 to give the intermediate 13 which undergoes aromatization by lose three molecules of secondary amines, under the reaction conditions, to give 14.
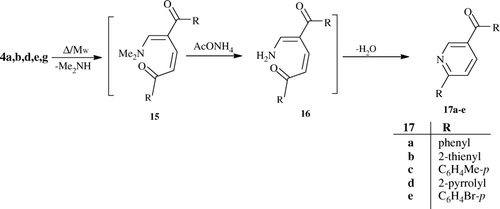
Heating enaminones 4a,b,d,e,g with excess of ammonium acetate, as an IL, at 110°C for 20 min or irradiating under MW for 1 min at 400 watt, 105°C yielded the 2-aryl (or heteroaryl)-5-aroyl (or hetroaroyl)-pyridines 17a–e. The formation of 17a–e may take place through an initial self-condensation of 4, via loss one molecule of dimethylamine, to yield the intermediate 15 which reacts directly with ammonium ion to give a further enamine intermediate 16. The latter undergoes intramolecular cyclocondensation, via loss one molecule of water, to give the final products 17a–e ().
Temperature measured during exposure in MW experiment which confirms the rate augmentation during MW heating. Temperature measurement is performed for all reactions with thermocouple sensors, which show sufficient accuracy for the presented reactions used to monitor the temperature inside the vessel; it was found that ≈105–110°C, to ensure reproducibility.
Experimental
All melting points were measured on a Gallenkamp electrothermal melting point apparatus and are uncorrected. The IR absorption spectra were measured on a Nicolet Magna 520FT IR spectrophotometer. 1H NMR and 13C NMR spectra were recorded in deuterated dimethylsulfoxide [DMSO] or deutrated chloroform (CDCl3) at 200 MHz on a Varian Gemini NMR spectrometer and a Bruker DPX 400 MHz spectrometer using tetramethylsilane (TMS) as an internal reference. Mass spectra were performed on a Shimadzu GCMS-QP 1000 EX mass spectrometer at 70 eV. X-ray crystallography was carried out on a Kappa CCD Enraf Nonius FR 590 diffractometer, National Research Center, Dokki, Cairo, Egypt. MW irradiation was carried out using the commercial MW oven (SGO 1000 W), with a thermocouple used to monitor the temperature inside the vessel and it was found that ≈105–110°C. Elemental analyses were performed on Perkin Elmer 2400 CHN elemental analyzer flowchart. Elemental analyses (C, H, N, S) were conducted using the Elemental Analyser XBO and results were found to be within ±0.2% of the calculated values.
General procedure for the preparation of compounds 3a,b
Method I (Δ)
A mixture of pyridinium chloride ([PyH]Cl; 0.4 mol) and enamines 1a–f (0.1 mol) was heated at 110°C for 30–60 min and was allowed to cool to room temperature. Then, it was treated with ethanol. The solid product so formed was collected by filtration, dried, and recrystallized from ethanol.
Method II (MW)
A mixture of [PyH]Cl (0.4 mol) and enamines 1a–f (0.1 mol) was placed in the MW oven and irradiated at 400 watt, 105°C for 1–3 min. After cooling to room temperature, it was treated with ethanol. The solid product so formed was collected by filtration, dried, and recrystallized from ethanol.
Compound (3a, C9H3N3)
[55% (Method I), 67% (Method II)] as a brown powder; mp>300°C (from EtOH); ν max/(KBr)cm−1 3095 (aromatic CH), 2212 (CN); m/z (EI) 153 (M+, 43%). (Found: C, 70.55; H, 2.03; N, 27.43. C9H3N3 Calculated: C, 70.59; H, 1.97; N, 27.44%)
Compound (3b, C24H15N3O6)
[60% (Method I), 77% (Method II)] as a brown powder; mp>300°C; ν max/(KBr)cm−1 3089 (aromatic CH), 1510, 1341 (NO2); m/z (EI) 441 (M+, 100%); δH(400 MHz; DMSO-d 6 ) 7.68 (3 H, s, Ar-H), 8.10 (6 H, d, J 8, ArH), 8.58 (6 H, d, J 8, ArH). (Found: C, 65.39; H, 3.51; N, 9.63. C24H15N3O6 Calculated: C, 65.31; H, 3.43; N, 9.52%)
General procedure for the preparation of 1,3,5-Trisubstituted benzene 7a–j
Method I (Δ)
A mixture of [PyH]Cl (0.4 mol) and enaminones 4a–j (0.1 mol) was heated at 110°C for 20 min and was allowed to cool to room temperature. Then, it was treated with a mixture of ethanol/dioxane (3:1). The solid product so formed was collected by filtration, dried, and recrystallized from dioxane.
Method II (MW)
A mixture of [PyH]Cl (0.4 mol) and enaminones 4a–j (0.1 mol) was placed in the MW oven and irradiated at 400 Watt, 105°C for 1 min The reaction mixture left to cool to room temperature and then treated with a mixture of ethanol/dioxane (3:1). The solid product so formed was collected by filtration, dried, and recrystallized from dioxane.
(5-(3-Chlorobenzoyl)-1,3-phenylene)bis((4-chlorophenyl)methanone) (7c, C27H15Cl3O3)
Mp 184–186°C; ν max/(KBr)cm−1 3082 (aromatic CH), 1667 (C=O); m/z (EI) 492 (M+ +1, 14%); δH(400 MHz; DMSO-d6) 7.65 (6 H, d, J 8, ArH), 7.86 (6 H, d, J 8, ArH), 8.23 (3 H, s, Ar-H); δC(400 MHz; DMSO-d6) 129.46, 132.34, 135.36, 138.90 (C 6H4-Cl-p), 134.31, 137.81 (C 6H3-CO), 193.80(C=O). (Found: C, 65.70; H, 3.00. C27H15Cl3O3 Calculated: C, 65.68; H, 3.06%.)
(5-(3-Methylbenzoyl)-1,3-phenylene)bis(p-tolylmethanone) (7d, C30H24O3)
Mp 131–133°C; ν max/(KBr)cm−1 3035 (aromatic CH), 2925 (aliphatic CH), 1663 (C=O); m/z (EI) 432 (M+, 9%); δH(400 MHz; DMSO-d6) 2.40 (9 H, s, 3CH 3), 7.38 (6 H, d, J 8, ArH), 7.74 (6 H, d, J 8, ArH), 8.20 (3 H, s, Ar-H); δC(400 MHz; DMSO-d 6 ) 21.77 (3×CH3), 129.87, 130.64, 133.93, 144.46 (C 6H4-CH3-p), 134.08, 138.23 (C 6H3-CO), 194.48 (C=O). (Found: C, 83.40; H, 5.81. Calculated: C, 83.31; H, 5.59%.)
Benzene-1,3,5-triyltris((1H-pyrrol-2-yl)methanone) (7e, C21H15N3O3)
Mp 256–258°C; ν max/(KBr)cm−1 3287 (NH), 3050 (aromatic CH), 1669 (C=O); m/z (EI) 357 (M+, 31%); δH(400 MHz; DMSO-d6) 6.31 (3 H, t, J 2.2, 3×pyrrolyl H-4), 6.94 (3 H, d, J 2.2, 3×pyrrolyl H-3), 7.29 (3 H, d, J 2.2, 3×pyrrolyl H-5), 8.36 (3 H, s, Ar-H), 12.22 (3 H, s, pyrrole NH); δC(400 MHz; DMSO-d 6 ) 111.25, 120.39, 127.78, 130.75 (pyrrole carbons), 131.80, 139.42 (C 6H3-CO), 182.69 (3C=O). (Found: C, 70.66; H, 4.26; N, 11.66. C21H15N3O3 Calculated: C, 70.58; H, 4.23; N, 11.76%.)
Benzene-1,3,5-triyltris((4-bromophenyl)methanone) (7g, C27H15Br3O3)
Mp 205–207°C; ν max/(KBr)cm−1 3065 (aromatic CH), 1665 (C=O); m/z (EI) 626 (M+, 13%); 1H δH(400 MHz; DMSO-d6) 7.62-7.68 (12 H, m, ArH), 8.31 (3 H, s, Ar-H); δC(400 MHz; DMSO-d 6 ) 128.82, 131.58, 132.18, 135.02 (C 6H4-Br-p), 133.98, 138.10 (C 6H3-CO), 193.64 (C=O). (Found: C, 51.76; H, 2.32. C27H15Br3O3 Calculated: C, 51.71; H, 2.41%.)
Benzene-1,3,5-triyltris((4-nitrophenyl)methanone) (7j, C27H15N3O9)
Mp 196–198°C; ν max/(KBr)cm−1 3086 (aromatic CH), 1665 (C=O), 1526, 1337 (NO2); m/z (EI) 525 (M+, 12%); δH(400 MHz; DMSO-d6) 7.99 (6 H, d, J 8, ArH), 8.31 (6 H, d, J 8, ArH), 8.42 (3 H, s, Ar-H); δC(400 MHz; DMSO-d6) 123.93, 130.87, 140,98, 150.56 (C 6H4-NO2-p), 134.62, 137.74 (C 6H3-CO), 196.34 (C=O). (Found: C, 61.81; H, 2.79; N, 8.12. Calculated: C, 61.72; H, 2.88; N, 8.00%.)
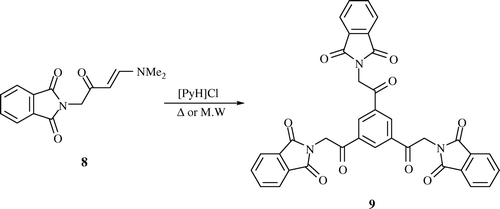
General procedure for the preparation of 2,2′,2″-(2,2′,2″-(Benzene-1,3,5-triyl)tris(2-oxoethane-2,1-diyl))triisoindoline-1,3-dione 9
Method I (Δ)
A mixture of [PyH]Cl (0.4 mol) and enaminone 8 (0.1 mol) was heated at 110°C for 20 min After cooling to room temperature, the reaction mixture was treated with ethanol. The solid product so formed was collected by filtration, dried, and recrystallized from ethanol.
Method II (MW)
A mixture of [PyH]Cl (0.4 mol) and enaminone 8 (0.1 mol) was placed in the MW oven and irradiated at 400 watt, 105°C for 0.5 min. After cooling to room temperature, it was treated with ethanol. The solid product so formed was collected by filtration, dried, and recrystallized from ethanol to give compound 9 as pale yellow crystals.
2,2′,2″-(2,2′,2″-(Benzene-1,3,5-triyl)tris(2-oxoethane-2,1-diyl))triisoindoline-1,3-dione (9, C36H21N3O9)
[89% (Method I), 98% (Method II)] as a brown powder; mp 295–297°C; ν max/(KBr)cm−1 3070 (aromatic CH), 1774 (C=O), 1704 (C=O); m/z (EI) 639 (M+, 7%); δH(400 MHz; DMSO-d6) 5.56 (6 H, s, 3CH2), 7.91-7.97 (12 H, m, phthalimide-H), 8.99 (3 H, t, J 8, Ar-H). (Found: C, 67.55; H, 3.49; N, 6.50. C36H21N3O9 Calculated: C, 67.61; H, 3.31; N, 6.57%.)
General procedure for the preparation of Triethyl benzene-1,3,5-tricarboxylate 14
Method I (Δ)
A mixture of [PyH]Cl (0.4 mol) and enaminoester 10a–c (0.1 mol) was heated at 110°C for 30 min and was allowed to cool to room temperature. Then, it was treated with ethanol. The solid product so formed was collected by filtration, dried, and recrystallized from ethanol.
Method II (MW)
A mixture of [PyH]Cl (0.4 mol) and enaminoester 10a–c (0.1 mol) was placed in the MW oven and irradiated at 400 watt, 105°C for 0.5 min. After cooling to room temperature, it was treated with ethanol. The solid product so formed was collected by filtration, dried, and recrystallized from ethanol, to give compound 14 as orange crystal.
Triethyl benzene-1,3,5-tricarboxylate (14, C15H18N6)
[55% (Method I), 83% (Method II)] as pale yellow; mp 127–129°C; ν max/(KBr)cm−1 3099 (aromatic CH), 2994 (aliphatic CH), 1715 (CO ester); m/z (EI) 294 (M+, 100%); δH(400 MHz; DMSO-d 6 ) 1.44 (9 H, t, J 6, 3×CH 3), 4.45 (6 H, q, J 6, 3×CH 2), 8.84 (3 H, s, Ar-H); δC(400 MHz; DMSO-d 6 ) 14.13 (3×CH3), 61.72 (3×CH2), 131.49, 135.30 (C 6H3-CO), 165.09 (C6H3-CO). (Found: C, 61.29; H, 6.14. C15H18N6 Calculated: C, 61.22; H, 6.16%.)
General procedure for the preparation of 2-Aryl-5-aroylpyridines 17a–e
Method I (Δ)
A mixture of ammonium acetate (0.4 mol) and enaminones 4a,b,d,e,g (0.1 mol) was heated at 110°C in an oil bath for 20 min and was allowed to cool to room temperature. Then, it was treated with ethanol. The solid product so formed was collected by filtration, dried, and recrystallized from ethanol.
Method II (MW)
A mixture of ammonium acetate (0.4 mol) and enaminones 4a,b,d,e,g (0.1 mol) was placed in the MW oven and irradiated at 400 watt, 105°C for 1 min. After cooling to room temperature, it was treated with ethanol. The solid product so formed was collected by filtration, dried, and recrystallized from ethanol.
Phenyl(6-phenylpyridin-3-yl)methanone (17a, C18H13NO)
[50% (Method I), 65% (Method II)] as a yellow crystal; mp 120–122°C; ν max/(KBr)cm−1 3080 (aromatic CH), 1662 (C=O); m/z (EI) 259 (M+, 86%); δH(400 MHz; DMSO-d 6 ) 7.20-8.01 (12 H, m, 10×Ar-H+2b×pyridine-H); 9.03 (1 H, s, pyridine H-6); δC(400 MHz; DMSO-d 6 ) 123.65, 137,79 (C 6H5), 123.87, 131.99 (COC 6H5), 135.01, 135.29, 167.90, 170.01 (pyridyl carbons), 189.99 (C=O). (Found: C, 83.24; H, 5.15; N, 5.48. Calculated: C, 83.37; H, 5.05; N, 5.40%.)
Thiophen-2-yl(6-(thiophen-2-yl)pyridin-3-yl)methanone (17b, C14H9NOS2)
[54% (Method I), 74% (Method II)] as a black powder; mp 128–129°C; ν max/(KBr)cm−1 3071 (aromatic CH), 1660 (C=O); m/z (EI) 271 (M+, 100%); δH(400 MHz; DMSO-d 6 ) 7.13 (1 H, t, J 4, C4 H 3S H-4), 7.32 (1 H, t, J 4, COC4 H 3S H-4), 7.22 (1 H, d, J 4, C4 H 3S H-3), 7.35 (1 H, d, J 4, C4 H 3S H-5), 7.71 (1 H, d, J 4, COC4 H 3S H-3), 7.77 (1 H, d, J 4, COC4 H 3S H-5), 8.11 (1 H, d, pyridine, H-3), 8.19 (1 H, d, pyridine H-4), 8.99 (1 H, s, pyridine H-6). (Found: C, 61.85; H, 3.34; N, 5.09. C14H9NOS2 Calculated: C, 61.97; H, 3.34; N, 5.16%.)
p-Tolyl(6-p-tolylpyridin-3-yl)methanone (17c, C20H17NO)
[62% (Method I), 75% (Method II)] as an orange crystal; mp 116–118°C; ν max/(KBr)cm−1 3090 (aromatic CH), 2915 (aliphatic CH), 1667 (C=O); m/z (EI) 287 (M+, 86%); δH(400 MHz; DMSO-d 6 ) 7.23 (2 H, d, J 8, ArH), 7.40 (2 H, d, J 8, ArH), 7.71 (2 H, d, J 8, ArH), 7.79 (2 H, d, J 8, ArH), 8.09-8.13 (2 H, m, pyridine H-3 and H-4), 8.93 (1 H, s, pyridine H-6); δC(400 MHz; DMSO-d 6 ) 21.55 (2×CH3), 127.48, 129.27, 132.22, 134.50 (C 6H4-CH3-p), 129.29, 130.11, 141.20 (COC 6H4-CH3-p), 120.11, 138.07, 154.53, 161.09 (pyridine carbons), 186.01 (C=O). (Found: C, 83.68; H, 5.83; N, 4.82. C20H17NO Calculated: C, 83.59; H, 5.96; N, 4.87%.)
(6-(1H-Pyrrol-2-yl)pyridin-3-yl)(1H-pyrrol-2-yl)methanone (17d, C14H11N3O)
[50% (Method I), 66% (Method II)] as a brown powder; mp 133–134°C; ν max/(KBr)cm−1 3086 (aromatic CH), 1670 (C=O) 3353 (NH); m/z (EI) 237 (M+, 22%); δH(400 MHz; DMSO-d 6 ) 6.38 (1 H, t, J 2.2, pyrrole H-4), 6.41 (1 H, t, J 2.2, pyrrole H-4), 6.75 (1 H, d, J 2.2, pyrrole H-3), 7.15 (1 H, d, J 2.2, pyrrole H-3), 7.22 (1 H, d, J 2.2, pyrrole H-5), 7.47 (1 H, d, J 2.2, pyrrole H-5), 7.62 (1 H, d, J 8, pyridine H-3), 8.58 (1 H, d, J 8, pyridine H-4), 9.10 (1 H, s, pyridine H-6), 11.22 (2 H, s, pyrrole NH). (Found: C, 70.93; H, 4.56; N, 17.64. C14H11N3O Calculated: C, 70.87; H, 4.67; N, 17.71%.)
(4-Bromophenyl)(6-(4-bromophenyl)pyridin-3-yl)methanone (17e, C18H11Br2NO)
[55% (Method I), 77% (Method II)] as an orange crystal; mp 192–194°C; ν max/(KBr)cm−1 3095 (aromatic CH), 1668 (C=O); m/z (EI) 417 (M+, 45%); δH(400 MHz; DMSO-d 6 ) 7.44 (2 H, d, J 8, ArH), 7.62 (2 H, d, J 8, ArH), 7.76 (2 H, d, J 8, ArH), 7.83 (2 H, d, J 8, ArH), 7.97 (1 H, d, pyridine H-3), 8.18 (1 H, d, J 8, pyridine H-4), 8.98 (1 H, s, pyridine H-6). (Found: C, 51.96; H, 2.63; N, 3.45. C18H11Br2NO Calculated: C, 51.83; H, 2.66; N, 3.36%.)
Notes
1. Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary puplications No. CCDC 686291. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; Email: [email protected]).
Related Research Data
References
- Wang , J. ; Fan , X. ; Zhang , X. ; Han , L. Can. J. Chem . 2004 , 82 , 1192 1196 .
- Qu , G. ; Han , S. ; Zhang , Z. ; Geng , M. ; Xue , F. Can. J. Chem . 2006 , 84 , 819 824 .
- Al-Zaydi , K.M. ; Borik , R.M. Molecules 2007 , 12 , 2061 2079 .
- Al-Saleh , B. ; Makhseed , S. ; Hassaneen , H.M.E. ; Elnagdi , M.H. Synthesis 2006 , 59 62 .
- Hassaneen , H.M.E. ; Hassaneen , H.M. ; Elnagdi , M.H. Z. Naturforsch. B: Chem. Sci . 2004 , 59 , 1132 1136 .
- Ghozlan , S.A.S. ; Abdelhamid , I.A. ; Gaber , H. ; Elnagdi , M.H. J. Heterocyclic Chem . 2005 , 42 , 1185 1189 .
- Al-Mousawi , S.M. ; EL-Apasery , M.A. ; Elnagdi , M.H. Heterocycles 2008 , 75 , 1151 1161 .
- (a) Al-Zaydi , K.M. ; Borik , R.M. ; Elnagdi , M.H. J. Heterocyclic Chem . 2007 , 44 , 1187 1189 (b) Al-Zaydi K.M. Borik R.M. Elnagdi M.H. Ultrason. Sonochem. 2009 16 660 668 (c) Al-Zaydi K.M. Borik R.M. Elnagdi M.H. 2009 16 805 809
- Al-Awadi , N.A. ; Abdelkhalik , M.M. ; Abdelhamid , I.A. ; Elnagdi , M.H. SynLett 2007 , 2979 2982 .
- Al-Zaydi , K.M. ; Al-Shamary , A. ; Elnagdi , M.H. J. Chem. Res . 2006 , 408 411 .
- Al-Zaydi , K.M. ; Al-Shamary , A. Oriental J. Chem . 2007 , 23 , 387 391 .
- AbdelKhalik , M.M. ; Elnagdi , M.H. Synth. Commun . 2002 , 32 , 159 164 .
- Alzaydi , K.M. Molecules 2003 , 8 , 541 555 .
- Almazroa , S. ; Elnagdi , M.H. ; Salah El-Din , A.M. J. Heterocyclic Chem . 2004 , 41 , 267 272 .
- Tundo , P. ; Anastas , P. ; StC Black , D. ; Breen , J. ; Collins , T. ; Memoli , S. ; Miyamoto , J. ; Polyakoff , M. ; Tumas , W. Pure Appl. Chem . 2000 , 72 , 1207 1212 .
- Hoz , A. ; Diaz-Ortiz , A. ; Moreno , A. Chem. Soc. Rev . 2005 , 34 , 164 178 .
- Lidstrom , P. ; Tierney , J. ; Wathey , B. ; Westman , J. Tetrahedron 2001 , 57 , 9225 9283 .
- (a) Al-Omran , F. ; El-Khair , A.A. J. Chem. Res . 2006 , 6 9 (b) Al-Omran F. El-Khair A.A. J. Heterocyclic Chem. 2005 42 307 312
- Kascheres , C.M. Braz. Chem. Soc . 2003 , 14 , 945 969 .
- Greenhill , J.V. Chem. Soc. Rev . 1977 , 6 , 277 294 .
- Zhuo , J.C. ; Schenk , K. Helve Chim. Act . 1997 , 80 , 2137 2147 .
- Al-Mousawi , S. ; John , E. ; Al-Kandery , N. J. Heterocyclic Chem . 2004 , 41 , 381 385 .
- Migeon , H. ; Fradet , A. ; Madec , P.J. ; Marechal , E. Bul. Soc. Chim. (Fr) 1995 , 132 , 967 971 .
- Alzaydi , K.M. ; Hafez , E.A. J. Chem. Res (s) 1999 , 360 361 .