Abstract
Synthesis of structurally varied β-arylvinyl bromides has been accomplished in an eco-friendly “On-water” condition through bromodecarboxylation reaction of appropriately substituted cinnamic acids with potassium bromide using hydrogen peroxide as oxidant and sodium tungstate as catalyst. Salient mechanistic features have also been highlighted.
Introduction
Substituted β-arylvinyl bromides act as extremely versatile precursors for the Pd(0) Citation1 and Ni(0) Citation2 mediated syntheses of various diene and eneyne systems, α,β-unsaturated aldehydes Citation3, and α,β-unsaturated carboxylic acids Citation4 as well as in CuI-L-proline-mediated reactions with active methylene compounds for the synthesis of α-methylene enones Citation5, Ullmann reaction with imidazole in ionic liquid Citation6, enantioselective synthesis of (+) (S,S)-reboxetine Citation7, stereoselective synthesis of vinyl selenide using catalytic amount of CuI in [bmIm]BF4 Citation8, construction of furan moiety by Suzuki coupling Citation9, and stereocontrolled synthesis of α,α-difluorosubstituted phenyl butenoate Citation10. (E)-β-arylvinyl bromides were classically prepared by Hunsdiecker reaction Citation11–13. With time, numerous protocols have been developed, for example, Pd(0)-mediated coupling of trans-1,2-bis-(tri-n-butylstannyl)-ethylene with aryl bromides Citation14, halodecarboxylation of α,β-unsaturated carboxylic acids with a variety of reagents, namely, N-bromosuccinimide (NBS) and iodosylbenzene Citation15, NBS and Mn(III)-acetate Citation16, NBS in the presence of tetrabutylammonium trifluoroacetate Citation17, potassium bromide–hydrogen peroxide in the presence of sodium molybdate Citation18, LiBr-CAN Citation19, selectofluor Citation20, molybdate-exchanged Mg–Al-LDH Citation21, and diphosphorous tetraiodide in the presence of tetraethylammonium bromide in carbon disulfide Citation22; reductive debromination of gem-dibromides with indium in the presence of ammonium chloride in aqueous ethanolic medium Citation23, microwave-mediated base-catalyzed reduction of 1,1-dibromoalkenes with diethyl phosphonate (24a), silver acetate-promoted dehalodecarboxylation of 3-aryl-2,3-dibromopropanoic acid (24b), samarium-mediated reduction of 1,1-dibromoalkenes Citation25, and dephosphorylation of α,β-unsaturated phosphonic acids with bis-collidine-bromonium hexafluorophosphate Citation26. Many of them involve the use of exotic substrates (23, 24a, 25, 26), toxic organic solvents Citation22, expensive reagents Citation14 Citation15 Citation17 Citation20–26 often associated with considerable toxicity (14–17, 22, 24a, 26), drastic reaction conditions, long reaction time Citation17 Citation22, and low yield of the products Citation15 Citation17 Citation19 Citation20 Citation25 , sometimes as a mixture of two diastereomers Citation20 Citation25 or contaminated with other by-products Citation15 Citation25. Reduction of the use of hazardous chemicals and reagents is one of the most important challenges Citation27 in the present scenario to minimize pollution and the risks associated with the production and utilization of chemicals. Water is a promising choice Citation28–34 as the medium of organic reactions from the standpoint of sustainability and chemical safety. From the environmental perspective, hydrogen peroxide is a good choice as oxidant which was used for eco-friendly oxidation of olefins, alcohols, and sulfides Citation35, α-bromination of active methylene compounds with KBr in HCl Citation36, hydroxylation of haloarenes and heterohaloarenes Citation37, free radical bromination in aqueous HBr Citation38, and oxidation of alkenes–arenes to ketones in aqueous HBr Citation39 . Oxidative property of H2O2 can be amplified by use of other catalysts in various transformations, e.g. α-bromination of active methylene compounds using V2O5–NH4Br system Citation40, epoxidation of unsaturated terpenes with Na2WO4 Citation41, electrophilic bromination across olefin, acetylene, and ring bromination of aromatic rings using KBr, HBr, and NH4VO3 Citation42. Inspired by the above literature precedence, we have ventured to explore the catalytic activity of relatively less-toxic Na2WO4 for the eco-compatible synthesis of (E)-vinyl bromides in aqueous medium using KBr as a non-toxic brominating agent and H2O2 as an eco-friendly oxidant.
Results and discussion
The α,β-unsaturated carboxylic acids Citation43 Citation44 were treated with an aqueous solution of KBr containing 30% H2O2 in the presence of catalytic amount of Na2WO4·2H2O, as presented in . Detailed results have been presented in .
Scheme 1. “On-water’’ synthesis of E-vinyl bromides from β-aryl-α, β-eneoic acids.
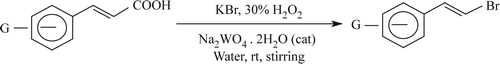
Table 1. Synthesis of (E)-vinyl bromides from aromatic α,β-eneoic acids.
According to the , several α,β-unsaturated carboxylic acids with varied substituents in the aromatic ring underwent clean bromodecarboxylation through operationally simple procedure using easily accessible and eco-friendly reagents to the corresponding (E)-β-arylvinyl bromides in good yields without formation of side products. The desired product was obtained in almost pure form within short reaction time and well characterized by spectroscopic techniques. The conversion of unsaturated acid to the corresponding vinyl bromides was ascertained by the appearance of two characteristic doublets at ▵ 6.78 and ▵ 7.12 having J=nearly 14.0 Hz indicating trans configuration the olefinic double bond. It has been found that reactions of cinnamic acids bearing electron-donating groups (Entries 2, 4, and 5) are more rapid and give high yield of products than those bearing electron-withdrawing groups (Entry 3). Cinnamic acid with an electron-donating group at para-position is more susceptible to react with KBr than at ortho position (Entry 6 versus Entry 4). Acid-labile methylenedioxy moiety survived under the said reaction condition (Entry 5). It has been observed that the cinnamic acids having a strong electron-withdrawing substituent remained totally unaffected in the present reaction condition (Entries 8–10). Like α,β-enoic acid, α,β-yneoic acid produced arylalkynyl bromide (Entry 11). Introduction of a substituent at α-carbon led to the formation of the corresponding β-bromostyrene with a satisfactory yield (Entries 14 and 15). However, γ-aryl γ-keto-α,β-unsaturated acid (bearing a keto group exerting both –R and –I effects between the aromatic ring and the double bond) did not produce the desired vinyl bromide even after a prolonged time (Entry 12) and it was recovered unchanged.
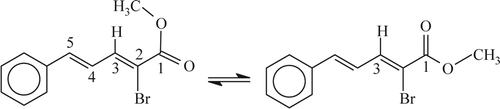
Interestingly, 2-cinnamyledenemalonic acid (Entry 13) furnished 2-bromo-2-cinnamyledene acetic acid where the carboxylic group cis with respect to diene moiety underwent decarboxylation remaining the other COOH intact, as evident from the singlet at δ 3.87 due to carbomethoxy protons in its methyl ester. Z-configuration of the C = C linkage between C-2 and C-3 has been established by 2D nOe experiments (done in 500 MHz NMR using mixing time 0.3 seconds at 27°C). The cross-peaks correlate between δ 7.84 and δ 3.87, the distance between C-3H and carbomethoxy protons has been estimated to be <5 Å. The nOe between C-3H and methyl ester protons is not very strong, because these protons are not always very close due to conformational equilibrium ().
Figure 1. Conformational equilibrium of methyl Z-2-bromo-5-phenylpent-2, 4-dienoate.
The present bromodecarboxylation reaction takes place in water which is the most eco-compatible medium. Cinnamic acid, being sparingly soluble in water (solubility in water is 3.34×10–3 mol L–1), remains mainly as a suspension in the aqueous reaction mixture with only 1.34% being in solution. Therefore, the present bromodecarboxylation reaction definitely takes place under “On-water” Citation29 condition. The said reactions were carried out using water-miscible organic co-solvents as additives to increase the miscibility of the sparingly soluble organic substrate, namely, cinnamic acid, with aqueous reaction mixture (). As evident in , increased miscibility has been found detrimental for the extent of conversion. Therefore, the efficacy of the present “On-water” protocol has been firmly established.
Table 2. Sodium tungstate-catalyzed bromodecarboxylation of cinnamic acid in aqueous medium in the presence of water-miscible co-solvents.
Use of sodium tungstate (which is practically safe Citation45 bearing very little toxicity) in catalytic amount along with hydrogen peroxide as an eco-friendly oxidant (by-product is water), potassium bromide as a non-toxic brominating agent in the most environmentally accepted medium, namely water, altogether makes the present protocol a greener and more sustainable alternative to many of the existing methods for the synthesis of β-arylvinyl bromides. In this protocol, waste production is extremely small. Ethyl acetate, used for extraction, is a relatively eco-compatible organic solvent Citation46 and can be recycled. Therefore, the present “On-water” protocol minimizes the dispersal of harmful chemicals in the environment and maximizes the use of renewable resource.
Cinnamic acid remained totally unaffected in the presence of aqueous KBr and 30% H2O2 in the absence of Na2WO4·2H2O clearly demonstrating a crucial role of Na2WO4 in this bromodecarboxylation reaction. Aliphatic α,β-unsaturated carboxylic acid (e.g. crotonic acid) remained unaffected, which proved the necessity of aromatic ring. Phenylacetic acid was recovered unchanged which eliminates the possibility of the radical pathway (which is the most accepted pathway for the classical Hunsdiecker reaction involving silver carboxylate). The solution of Na2WO4 (0.3 mmol) and KBr (2 mmol) in 2 mL water is basic in nature (pH 11.70). Addition of solid cinnamic acid (1 mmol) changed the pH to 7.92. When H2O2 (2 mL) was added, the pH became 5.99. The pH of the medium became 7.74 at the end of the reaction. Sodium salt of cinnamic acid remained unaffected and did not afford vinyl bromide, indicating the necessity of the acidic hydrogen in the COOH moiety. Again, cinnamic acid and ethyl cinnamate, on reaction with HBr, Na2WO4·2H2O, and 30% H2O2 in aqueous medium-furnished dibromocinnamic acid and ethyl dibromocinnamate, respectively. But ethyl cinnamate remained unchanged on treatment with aqueous KBr, Na2WO4·2H2O, and 30% H2O2. So, the requirement of COOH moiety for this transformation was further substantiated.
From the above observations, it is very clear that acidity is a crucial factor for this reaction and only a trace amount of acid is required for bromodecarboxylation reaction, where the weakly acidic COOH group of the substrate acts as the proton source. In the medium of higher acidity, considerable amount of molecular bromine might be generated (evident from dark-yellow coloration) for normal electrophilic addition reaction. In the presence of a weak organic acid, KBr, 30% H2O2, and catalytic amount of Na2WO4·2H2O, the concentration of bromine is not sufficient to produce molecular bromine to be utilized in the addition of Br2 across double bond of cinnamic acid or cinnamate ester. This was supported by the fact that anisole remained totally unaffected on treatment with aqueous KBr, 30% H2O2, and catalytic amount of Na2WO4·2H2O, but furnished a mixture of 4-bromo and 2,4-dibromo anisole on treatment with 30% H2O2 in the presence of HBr in place of KBr (in the presence as well as in the absence of Na2WO4). Therefore, the amount of electrophilic brominating agent was insufficient in the medium of lower acidity to promote any electrophilic substitution reaction, but in the medium of higher acidity substantial amount of “electrophilic Br” has been generated to promote normal bromination reaction, depending on the nature of the substrates. On the basis of the aforesaid observations, a plausible mechanism has been proposed in .
Figure 2. Catalytic role of sodium tetraperoxotungstate.
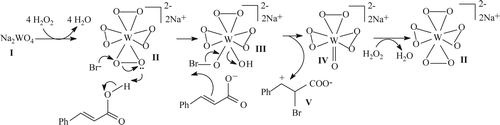
At the outset, sodium tungstate (I) reacts with H2O2 to form tetraperoxotungstate (II) Citation49. Protonation by cinnamic acid and subsequent cleavage of the one of the peroxo rings by concomitant nucleophilic attack of Br– produces III, where Br– is formally oxidized to an equivalent of the Br+ species. This is why the presence of COOH moiety is essential and COO– fails to react. III is converted to IV by nucleophilic attack of the double bond of the cinnamate anion on the electrophilic “Br” of III. As a result, cyclic bromonium ion is transiently formed and rapidly converted to a carbocationic species V, where the +ve charge is best accommodated on the carbon bearing aromatic ring. Low yield for 2-methoxycinnamic acid might be due to steric crowding causing increase of steric strain in the bromonium ion. As a discrete carbocationic intermediate is presumably involved in this reaction, therefore, stabilization through conjugation with the adjacent benzene ring becomes a crucial factor for its formation and subsequent reactions. Presence of an electron-withdrawing group in the aromatic ring (Entries 8, 9 and 10) as well on the carbon bearing +ve charge (Entry 12) destabilizes the intermediate carbocation, so the reactions, in these cases, do not take place. II is regenerated from IV with H2O2 Citation49 and enters the catalytic cycle again.
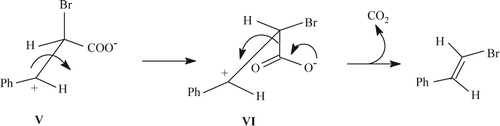
The carbocationic intermediate V then undergoes rotation about C–C bond to form VI, where the COO– moiety is antiperiplanar with respect to the vacant p-orbital on the cationic center satisfying the stereoelectronic requirement for decarboxylative elimination. As a consequence, the geometry of the newly formed olefinic linkage has been found trans ().
Figure 3. Mechanistic explanation for the trans-stereoselectivity of bromodecarboxylation.
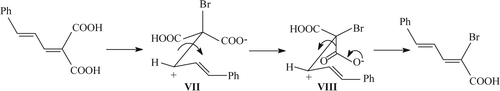
In case of 2-cinnamylidenemalonic acid (Entry 13 in ), that COOH is preferentially deprotonated and subsequently decarboxylated (VII–VIII) which, in order to satisfy the stereoelectronic requirement, needs to overcome lower rotational barrier putting the bulky substituents (namely, cinnamylidene and carboxylic acid) away from each other around the incipient double bond ().
Figure 4. Preferred decarboxylation of 2-cinnamylidenemalonic acid.
Experimental
General procedure for preparation of (E)-β-arylvinyl bromides
A suspension of α,β-unsaturated aromatic carboxylic acids (2 mmol) was stirred with a solution of potassium bromide (4 mmol, SRL, India) and sodium tungstate dihydrate (0.6 mmol, E. Merck, India) in water (4 mL). Then 30% hydrogen peroxide (4 mL, sd-Fine-Chemicals Private Limited, India) was added dropwise to it at room temperature with vigorous stirring. A rapid reaction took place and the temperature rose to 60°C. The reaction mixture was allowed to cool to room temperature and vigorously stirred till completion of reaction (monitored by thin layer chromatography on pre-coated silica gel plates, E. Merck, Germany) and the product was extracted from aqueous layer using ethyl acetate (3×10 mL). The combined organic layer was washed successively with water, aqueous NaHCO3, and brine; dried over anhydrous sodium sulfate and the solvent was evaporated under reduced pressure to get the vinyl bromide as the product in practically pure form. The product was further purified by filtration chromatography on a short column of silica gel, if needed.
Conclusion
A simple and efficient method for oxidative bromodecarboxylation of α,β-unsaturated aromatic carboxylic acids have been developed using aqueous KBr and 30% H2O2 mediated by Na2WO4·2H2O. There are significant improvements offered by the present method over existing procedures which include: (a) no undesired side reaction; (b) mild conditions to tolerate several sensitive moieties; (c) good stereoselectivity; (d) high yields; and (e) environmental friendly reaction conditions by using water as reaction medium and comparatively eco-compatible reagents and organic solvent. This present protocol not only demonstrates the efficacy of “On-water” reactions from the preparative standpoints, but also provides a practical, greener, and more sustainable alternative to the existing methodologies for the stereoselective synthesis of E-β-arylvinyl bromides from easily accessible substrates in an eco-friendly condition using reagents of extremely little toxicity.
Supplementary Data
Download Zip (10 KB)Acknowledgements
The authors express profound thanks to Mr. Samit Kumar Dutta of Indian Institute of Chemical Technology, Hyderabad, for his invaluable help in course of this investigation. Infrastructural support from DST-FIST program, financial assistance from UGC-CAS-program in Chemistry, Jadavpur University and DST-PURSE program are also gratefully acknowledged.
Supplementary information: The spectral characterization of the products described in has been furnished as the supplementary information.
Related Research Data
References
- Heck , R.F. 1985 . Palladium Reagents in Organic Synthesis , New York : Academic press .
- Tanaka , H. ; Kosaka , A. ; Yamashita , S. ; Morisaki , K. ; Torii , S. J. Org. Chem . 2000 , 65 , 4575 4583 .
- Brennfuhrer , A. ; Neumann , H. ; Klans , S. ; Riermeier , T. ; Almena , J. ; Beller , M. Tetrahedron 2007 , 63 , 6252 6258 .
- Xiaodan , Z. ; Howard , A. ; Zhengkum , Y. J. Org. Chem . 2006 , 71 , 3988 3990 .
- Pei , L. ; Qian , W. Synlett 2006 , 1719 1723 .
- Wang , Z. ; Bao , W. ; Jiang , Y. Chem. Comm . 2005 , 22 , 2849 2851 .
- Siddiqni , S.A. ; Narkhede , U.C. ; Lahoti , R.J. Synlett 2006 , 1771 1773 .
- Chang , D. ; Bao , W. Synlett 2006 , 1786 1788 .
- Tnan , D.T. ; Ting , D.T. ; Langer , P. Synlett 2006 , 2812 2814 .
- Ghattas , W. ; Hess , C.R. ; Iacazio , A. ; Hardre , A.R. ; Klinmann , J.P. ; Reghir , H. J. Org. Chem . 2006 , 71 , 8616 8621 .
- Johnson , R.G. ; Ingham , R.K. Chem. Rev . 1956 , 56 , 219 269 .
- Wilson , C.V. Org. React . 1957 , 9 , 332 387 .
- Berman , J.D. ; Price , C.C. J. Org. Chem . 1958 , 23 , 102 103 .
- Haack , R.A. ; Penning , T.D. ; Djuric , S.W. ; Dziuba , J.A. Tetrahedron Lett . 1988 , 29 , 2783 2786 .
- Anette , G. ; Karl , A.J. ; Staren , D. ; Andrzej , S. J. Org. Chem . 1994 , 59 , 3543 3546 .
- Chowdhury , S. ; Roy , S. Tetrahedron Lett . 1996 , 37 , 2623 2624 .
- Naskar , D. ; Chowdhury , S. ; Roy , S. Tetrahedron Lett . 1998 , 39 , 699 702 .
- Sinha , J. ; Layek , S. ; Mandal , C.G. ; Bhattacharjee , M. Chem. Comm . 2001 , 1916 1917 .
- Roy , S.C. ; Guin , C. ; Maiti , G. Tetrahedron Lett . 2001 , 42 , 9253 9255 .
- Ye , C. ; Shreeve , M.J. J. Org. Chem . 2004 , 69 , 8561 8563 .
- Choudary , B.M. ; Someshwar , T. ; Lakshmikantam , M. ; Reddy , C.V. Catal. Commun . 2004 , 5 , 215 219 .
- Telvekar , V.N. ; Chettiar , S.N. Tetrahedron Lett . 2007 , 48 , 4529 4532 .
- Ranu , B.C. ; Samanta , S. ; Guchhait , S.K. J. Org. Chem . 2001 , 66 , 4102 4103 .
- (a) Kuang , C. ; Senboku , H. ; Tokuda , M. Tetrahedron 2002 , 58 , 1491 1496 (b) Kuang, C.; Senboku, H.; Tokuda, M. Tetrahedron 2005, 61, 637–642.
- Wang , L. ; Li , P. ; Xie , Y. ; Ding , Y. Synlett 2003 , 8 , 1137 1139 .
- Hind , L. ; Sylvie , R. ; Gerard , R. Tetrahedron Lett . 2005 , 46 , 1635 1637 .
- Djerassi , C. Angew. Chem. Int. Ed . 2004 , 43 , 2330 2332 .
- Leadbeater , N.E. Chem. Commun . 2005 , 2881 2902 .
- Narayan , S. ; Muldoon , J. ; Finn , M. ; Fokin , G.V.V. ; Kolb , H.C. ; Sharpless , K.B. Angew. Chem. Int. Ed . 2005 , 44 , 3275 3279 .
- Carril , M. ; SanMartin , R. ; Tellitu ; I. ; Dominguez , E. Org. Lett . 2006 , 8 , 1467 1470 .
- Butler , R.N. ; Coyne , A.G. ; Molony , E.M. Tetrahedron Lett . 2007 , 48 , 3501 3503 .
- Zhang , Q.Y. ; Liu , B-K. ; Chen , W-Q. ; Wu , Q. ; Lin , X-F. Green Chem . 2008 , 10 , 972 977 .
- Lipshutz , B.H. ; Chung , D.W. ; Rich , B. Org. Lett . 2008 , 10 , 3793 3796 .
- Ghassamipour , S. ; Sardarian , A.R. Tetrahedron Lett . 2009 , 50 , 514 519 .
- Noyori , R. ; Aoki , M. ; Kazuhiko , S. Chem. Comm . 2003 , 47 , 1977 1986 .
- Kirihara , M. ; Ugawa , S. ; Noguchi , T. ; Okubo , K. ; Monma , Y. ; Shimizu , I. ; Shimosaki , R. ; Hatono , A. ; Hirai , Y. Synlett 2006 , 2287 2289 .
- Cantrell , W.R. Jr Banta , W.E. ; Engles , T. Tetrahedron Lett . 2006 , 47 , 4249 4251 .
- Podgorsek , A. ; Stavber , S. ; Zupan , M. ; Iskra , J. Tetrahedron Lett . 2006 , 47 , 7245 7247 .
- Khan , A.T. ; Parvin , T. ; Choudhury , L.H. ; Ghosh , S. Tetrahedron Lett . 2008 , 48 , 2271 2274 .
- Khan , A.T. ; Goswami , P. ; Choudhury , L.H. Tetrahedron Lett . 2006 , 47 , 2751 2754 .
- Grigoropoulou , G. ; Clark , J.H. Tetrahedron Lett . 2006 , 47 , 4461 4463 .
- Moriuchi , T. ; Yamaguchi , M. ; Kikushima , K. ; Hirao , T. Tetrahedron Lett . 2007 , 48 , 2667 2670 .
- Saha , M. ; Roy , S. ; Chaudhuri , S.K. ; Bhar , S. Green Chem. Lett. Rev . 2008 , 1 , 113 121 .
- Vogel , A.I. Text Book of Practical Organic Chemistry , 5th ed , ELBS; Longman Scientific and Technical, Longman Group UK Ltd., Essex, England , 1998 1039 1041 .
- Material Safety Data Sheet . http://www.sciencelab.com (accessed Jan 5, 2010) .
- Alfonsi , K. ; Colberg , J. ; Dunn , P.J. ; Fevig , T. ; Jennings , S. ; Johnson , T.A. ; Peter Klein , H. ; Knight , C. ; Nagy , M.A. ; Perry , D.A. ; Stefaniak , M. Green Chem . 2008 , 10 , 31 36 .
- Kuang , C. ; Yang , Q. ; Senboku , H. ; Tokuda , M. Synthesis 2005 , 1319 1325 .
- Naskar , D. ; Roy , S. Tetrahedron 2000 , 56 , 1369 1377 .
- Hida , T. ; Nogusa , H. Tetrahedron 2009 , 65 , 270 274 .