Abstract
K2CO3–Al2O3 has been used as an efficient, eco-friendly, and cost-effective activating catalyst for the synthesis of 3-(2-chloroquinolin-3-yl)-1-substituted phenyl prop-2-en-1-ones starting from 2-chloroquinoline-3-carbaldehyde and acetophenone. The reactions were performed under ultrasound irradiations. A substantial enhancing effect in the yield was observed when the catalyst was activated under ultrasonic waves. The K2CO3–Al2O3 catalyst can compete with the traditional NaOH/MeOH system of catalysis when the reaction is carried out under ultrasound. Herein, a benign, eco-friendly, efficient, and extremely fast procedure for the synthesis of quinoline chalcones has been demonstrated successfully.
Keywords:
Introduction
The members of the chalcone and flavonoid family have attracted a great deal of interest due to their immense applications as anti-HIV agents Citation1, cytotoxic agents with antiangiogenic activity Citation2, antimalarials Citation3, and anti-inflammatory Citation4, antitumor Citation5, antibacterial, and anticancer pharmacological agents Citation6 Citation7. Chalcones are important intermediates in the synthesis of many pharmaceuticals. They are commonly synthesized via the Claisen-Schmidt condensation between acetophenone and benzaldehyde. This reaction is catalyzed by acids and bases under homogeneous conditions. Homogeneous reactions present several hurdles, such as catalyst recovery and waste disposal problems. In this respect, heterogeneous catalysts are considered as an ecofriendly alternative. The utilization of heterogeneous catalysts for the production of chalcones was reported in the literature Citation8–14, but there is no report of the use of K2CO3–Al2O3 as a catalyst in combination with ultrasound.
Exploring novel, better, cheap, safe, and environmentally friendly experimental techniques to carry out chemical transformations has been an important premise ofgr een synthesis.One such technique includes ultrasonic irradiation from ultrasonicator on reaction mixtures and on solid surfaces, which has emerged as a useful methodology for achieving better yields of the products, a significant reduction in reaction time, and a reduction or elimination ofen vironmentally detrimental solvents. For these reasons, ultrasonic assisted synthesis has clearly become a rapidly growing field of study especially for various organic transformations. Ultrasonic irradiation leads to the acceleration of numerous catalytic reactions as well as in homogeneous and heterogeneous systems Citation15. Furthermore, significant improvements can be realized with respect to the yields Citation16 Citation17. The sonochemical phenomena originate from the interaction between a suitable field of acoustic waves and a potentially reacting chemical system, and the interaction takes place through the intermediate phenomenon of acoustic cavitations. When one of the phases is a solid, the ultrasonic irradiation has several additional enhancement effects, and this is especially useful when the solid acts as a catalyst Citation18. In general, the sonication has beneficial effects on the chemical reactivity, such as accelerating the reaction, reducing the induction period, and enhancing the catalyst efficiency. Base-catalyzed processes such as Claisen-Schmidt condensation are commonly used for the manufacture of fine chemicals. Indeed, the development of environmentally friendly solid catalysts has recently experienced growing interest, and several review articles have been devoted to catalysis by solid bases, such as alkali-exchange zeolites, lithium nitrate Citation19, amino grafted zeolites Citation20, zinc oxide, silica-sulfuric acid Citation21, etc. Jhala et al. synthesized chalcones using basic alumina under microwave irradiation Citation22.
Keeping in view the advantages of ultrasonic irradiation and the use of chalcones as natural biocides, we have carried out the synthesis of some substituted quinoline chalcones by Claisen-Schmidt condensation in the present investigation. This reaction is generally carried out in presence of a base like NaOH or KOH; these are harmful, toxic, and polluting, and very low levels can produce irritation of the skin and eyes. Exposure to the solid or concentrated liquid can cause severe burns to the eyes, skin, and gastrointestinal tract, which may ultimately lead to death. This substance has been found in at least 49 of the 1,585 National Priorities List sites identified by the Environmental Protection Agency (EPA). Long-term exposure to sodium hydroxide in the air may lead to ulceration of the nasal passages and chronic skin irritation Citation23. Therefore, in the present investigation we have used K2CO3–Al2O3 as the condensing agent; it is cheap, non-toxic, and easy to use. We have incorporated alumina as a solid support in the reaction. Supported reagents on solid surfaces have been widely employed in organic synthesis. Reagents impregnated on solid materials present advantages over the conventional solution phase reactions, as good dispersion of active sites leads to improved reactivity and milder reaction conditions Citation24. Furthermore, the reaction can be easily carried out under solvent-free conditions under ultrasonic irradiation so as to minimize the pollution. Various substituted acetophenones were condensed with aromatic quinoline aldehydes in presence of K2CO3–Al2O3 to afford the desired chalcones in 85–90% yields under ultrasonic irradiations. The reaction was completed within 50–80 min. Thus, mild reaction conditions, a solvent-free protocol, ease of workup, high yields, stability, cost reduction, and a quick reaction time are features of this new protocol and provide the complete green modification to the classical synthesis of chalcones.
Results and discussion
Claisen-Schmidt condensation is a versatile method for the preparation of α, β-unsaturated carbonyl compounds (chalcones). The reaction is generally carried out in the presence of aqueous alkali. The concentration of the alkali generally lies between 10% and 60%. Other condensing agents that have been used for this reaction include alkali metal oxides, magnesium tert-butoxide Citation25, potassium carbon compounds (KC8) Citation26, boric anhydride Citation27, organo-cadmium compound, and lithium iodide Citation28, which are quite expensive and require a lot of precautions during their use. In the present investigation we have carried out the solvent-free condensation of 2-chloro-quinoline-3-carboxaldehyde and aromatic ketones in the presence of K2CO3–Al2O3 as shown in . The condensation was carried out in the presence of ultrasonic irradiation (solvent-free as well as with solvent) as well as with stirring for comparison. It was found that the condensation in the presence of ultrasonic irradiation worked well in solvent-free conditions in terms of yield and time of reaction execution. The condensation with stirring takes more time and gives a lower yield. In comparison to the previously reported condensing agents, this reagent is non-toxic, inexpensive, and easy to use. Furthermore, its use in combination with ultrasonic irradiation makes the process ecofriendly and economical and makes a new path in green chemical transformation. In comparison to the conventional method and reagents, the yields obtained are higher, as shown in . Moreover, no side products were observed in these reactions.
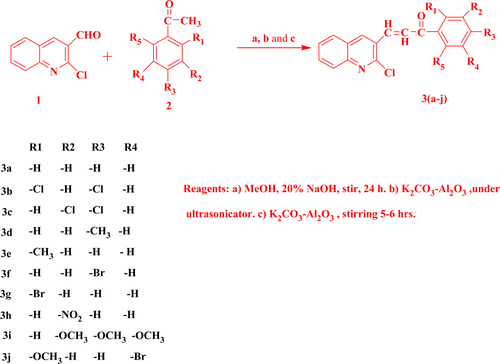
Table 1. Time taken and% yield for compounds 3a–3j.
The identity of the products obtained was confirmed on the basis of their elemental analysis and spectral data. The IR spectra of these compounds gave prominent peaks at 1740–1730 cm−1 (C=O), 3100–3000 cm−1 (C–H stret.), 1639–1641 cm−1 (CH=CH), and 853 cm−1 (Cl stret.). 1H NMR spectra of chalcones gave a doublet for vinylic protons around Δ7.53–8.31 and a multiplet for aromatic protons around Δ7.38–8.23. The mass spectra of these compounds gave molecular ion peaks corresponding to their molecular masses.
Experimental
All reagents, solvents, and catalyst are analytical grade from a commercial source and used directly. All the melting points were determined by the open tube capillaries method and are uncorrected. The purity of compounds was checked routinely by TLC (0.5 mm thickness) using silica gel-G coated Al-plates (Merck), and spots were visualized by exposing the dry plates in iodine vapors. IR spectra (νmax in cm-1) were recorded on a Schimadzu-IR Prestige 21 spectrophotometer using the KBr technique; 1H NMR spectra on a Bruker WM 400MHz NMR instrument using DMSO-d6 as solvent and TMS as an internal reference (chemical shifts in Δ, ppm); and mass spectra on a micromass Q-ToF high resolution mass spectrometer equipped with electrospray ionization (ESI) on a Masslynx 4.0 data acquisition system. The elemental analysis (C, H, N, and S) of compounds was performed on a Carlo Erba-1108 elemental analyzer. The results were found to be in good agreement with the calculated values. The ultrasonic assisted reactions were carried out in a Spectralab model UCB 40D ultrasonicator with a frequency of 40 kHz and a nominal power of 250 W. The reaction flask was located in the cleaner, where the surface of reactants is slightly lower than the level of the water.
Conventional method for synthesis of 3-(2-chloroquinolin-3-yl)-1-substituted phenyl prop-2-en-1-ones
To the solution of (0.01 mol) of 2-chloroquinoline-3-carbaldehyde in 5 mL of methanol, freshly prepared 2N methanolic NaOH solution (30 mL) was added in ice-cold condition and stirred for 10 min. To this (0.01 mol) of appropriate ketones was added, and the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was cooled in an ice bath and neutralized with dilute hydrochloric acid. The precipitate appeared and was separated by filtration and washed three times with 20 mL distilled water to give the crude product. The product so obtained was recrystallized from methanol. The purity of the products was checked using TLC (Merck silica gel 60F254) with a mixture of ethyl acetate and hexane as the mobile phase.
K2CO3–Al2O3 catalyzed general synthesis of 3-(2-chloroquinolin-3-yl)-1-substituted phenyl prop-2-en-1-ones
A mixture of (0.01 mol) of 2-chloroquinoline-3-carbaldehyde, acetophenone (0.01 mol), and potassium carbonate (0.08 g) was dissolved in dichloromethane (2 mL). The solution was adsorbed on neutral alumina (1.5 g) and air-dried. To the mixture 10 mL ethanol was added, and the mixture was stirred for 5–6 h. The progress of the reaction was monitored by TLC. After completion of the reaction excess ethanol was added and filtered to remove insoluble inorganic material. The filtrate was poured on crushed ice and neutralized with dilute HCl. Solid products were separated by filtration and recrystallized with ethanol to get the desired chalcones in 75–85% yields. The purity of the products was checked using TLC (Merck silica gel 60F254) with a mixture of ethyl acetate and hexane as the mobile phase.
Ultrasonic mediated K2CO3–Al2O3 catalyzed general synthesis of 3-(2-chloroquinolin-3-yl)-1-substituted phenyl prop-2-en-1-ones
The reaction was carried out in a Spectralab model UCB 40D ultrasonicator. A mixture of (0.01 mol) of 2-chloroquinoline-3-carbaldehyde, acetophenone (0.01 mol), and potassium carbonate (0.08 g) was dissolved in dichloromethane (2 mL). The solution was adsorbed on neutral alumina (1.5 g) and air-dried. The mixture was irradiated in the water bath of an ultrasonic cleaner for the period indicated in . The progress of the reaction was monitored by TLC. After completion of the reaction excess ethanol was added and filtered to remove insoluble inorganic material. The filtrate was poured on crushed ice and neutralized with dilute HCl. Solid products were separated by filtration and recrystallized with ethanol to get the desired chalcones in 80–90% yields. The purity of the products was checked using TLC (Merck silica gel 60F254) with a mixture of ethyl acetate and hexane as the mobile phase.
Yellow solid. Yield: 81%. R f=0.44 (EtOAc/hexane, 3:7). MP: 132–140°C. MS (M+): 293.062; FTIR (cm−1): 1732 (C=O), 1639 (CH=CH), 853 (C–Cl); 1H NMR (400 MHz, DMSO-d6) Δ/ppm: 7.53 (1H, d, Hα), 8.52 (1H, d, Hβ), 7.40–8.31 (m, 10H, aromatic). Anal. Calcd: C18H12ClNO: C, 73.54; H, 4.17; N, 4.75. Found: C, 73.60; H, 4.24; N, 4.78.
Data
3-(2-chloroquinolin-3-yl)-1-phenylprop-2-en-1-one (3a)
Prepared by the preceding method from 1 (0.01 mol) and acetophenone (0.01 mol). Yellow solid. Yield: 81%. R f=0.44 (EtOAc/hexane, 3:7). MP: 132–140°C. MS (M+): 293.06 (100%), 295.02 (33%). FTIR (cm−1): 1732 (C=O), 1639 (CH=CH), 853 (C–Cl). 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.53 (1H, d, Hα), 8.52 (1H, d, Hβ), 7.40–8.3 (m, 1H, 2H, 3H, 4H, 5H, 1′H, 2′H, 3′H, 4′H, 5′H). Anal. Calcd: C18H12ClNO: C, 73.54; H, 4.17; N, 4.75. Found: C, 73.60; H, 4.24; N, 4.78.
3-(2-chloroquinolin-3-yl)-1-(2, 4-dichoro phenyl)-)-prop-2-en-1-one (3b)
Prepared by the preceding method from 1 (0.01 mol) and 2, 4-dichloroacetophenone (0.01 mol). Pale yellow solid. Yield: 87%. R f=0.65 (EtOAc/hexane, 3:7). MP: 135–138°C; MS (M+): 360.98 (100%), 362.74 (95%). FTIR (cm−1): 1732 (C=O), 1639 (CH=CH), 853 (C–Cl). 1H NMR (400 MHz, DMSO-d6) Δ/ppm: 7.65 (1H, d, Hα), 8.51 (1H, d, Hβ), 7.34–8.32 (m, 1H, 2H, 3H, 4H, 5H, 2′H, 4′H, 5′H). Anal. Calcd: C18H10Cl3NO: C, 59.60; H, 2.76; N, 3.81. Found: C, 59.65; H, 2.79; N, 3.86.
3-(2-chloroquinolin-3-yl)-1-(3, 4-dichloro phenyl)-prop-2-en-1-one (3c)
Prepared by the preceding method from 1 (0.01 mol) and 3, 4-dichloroacetophenone (0.01 mol). Pale yellow solid. Yield: 83%. R f=0.61 (EtOAc/hexane, 3:7). MP: 145–148°C; MS (M+): 360.21 (100%), 362.76 (96%); FTIR (cm−1): 1730 (C=O), 1643 (CH=CH), 853 (C–Cl); 1H NMR (400 MHz, DMSO-d6) Δ/ppm: 7.34 (1H, d, Hα), 8.55 (1H, d, Hβ), 7.54–8.25 (m, 1H, 2H, 3H, 4H, 5H, 1′H, 4′H, 5′H). Anal. Calcd: C18H10Cl3NO: C, 59.40; H, 2.76; N, 3.80. Found: C, 59.45; H, 2.78; N, 3.88.
3-(2-chloroquinolin-3-yl)-1-(4-methyl phenyl)-prop-2-en-1-one (3d)
Prepared by the preceding method from 1 (0.01 mol) and 4-methylacetophenone (0.01 mol). Yellow crystalline solid. Yield: 80%, R f=0.56 (EtOAc/hexane, 3:7). MP: 140–148°C. MS (M+): 307.18 (100%), 309.09 (32%). FTIR (cm−1): 1728 (C=O), 1643 (CH=CH), 853 (C–Cl). 1H NMR (400 MHz, DMSO-d6) Δ/ppm: 2.38 (3H, s, CH3), 7.54 (1H, d, Hα), 8.59 (1H, d, Hβ), 7.29–8.33 (m, 1H, 2H, 3H, 4H, 5H, 1′H, 2′H, 4′H, 5′H). Anal. Calcd: C19H14ClNO: C, 74.12; H, 4.56; N, 4.49. Found: C, 74.15; H, 4.58; N, 4.18.
3-(2-chloroquinolin-3-yl)-1-(2-methyl phenyl)-prop-2-en-1-one (3e)
Prepared by the preceding method from 1 (0.01 mol) and 2-methylacetophenone (0.01 mol). Yellow crystalline solid. Yield: 85%. R f=0.51 (EtOAc /hexane, 3:7). MP: 132–140°C. MS (M+): 307.16 (100%), 309.04 (32%). FTIR (cm−1):1728 (C=O), 1643 (CH=CH), 853 (C–Cl). 1H NMR (400 MHz, DMSO-d6) Δ/ppm: 2.38 (3H, s, CH3), 7.54 (1H, d, Hα), 8.51 (1H, d, Hβ), 7.29–8.27 (m, 1H, 2H, 3H, 4H, 5H, 2′H, 3′H, 4′H, 5′H). Anal. Calcd: C19H14ClNO: C, 74.11; H, 4.56; N, 4.49. Found: C, 74.15; H, 4.58; N, 4.18.
3-(2-chloroquinolin-3-yl)-1-(4-bromophenyl) prop-2-en-1-one (3f)
Prepared by the preceding method from 1 (0.01 mol) and 4-bromoacetophenone (0.01 mol). Pale yellow crystalline solid. Yield: 84%. R f=0.59 (EtOAc/hexane, 3:7). MP: 146–150°C. MS (M+): 370.13 (100%), 372.54 (77%). FTIR (cm−1): 1728 (C=O), 1643 (CH=CH), 853 (C–Cl), 588 (C–Br). 1H NMR (400 MHz, DMSO-d6) Δ/ppm: 7.54 (1H, d, Hα), 8.53 (1H, d, Hβ), 7.43–8.31 (m, 1H, 2H, 3H, 4H, 5H, 1'H, 2'H, 4'H, 5'H). Anal. Calcd: C18H11ClBrNO: C, 58.01; H, 2.96; N, 3.74. Found: C, 58.04; H, 2.98; N, 3.78.
3-(2-chloroquinolin-3-yl)-1-(2-bromo phenyl)-prop-2-en-1-one (3g)
Prepared by the preceding method from 1 (0.01 mol) and 2-bromoacetophenone (0.01 mol). Yellow crystalline solid. Yield: 86%. R f=0.58 (EtOAc/hexane, 3:7). MP: 166–170°C; MS (M+): 370.19 (100%), 372.77 (77%). FTIR (cm−1): 1728 (C=O), 1643 (CH=CH), 853 (C–Cl), 588 (C–Br); 1H NMR (400 MHz, DMSO-d6) Δ/ppm: 7.54 (1H, d, Hα), 7.90 (1H, d, Hβ), 7.43–8.32 (m, 1H, 2H, 3H, 4H, 5H, 2′H, 3′H, 4′H, 5′H). Anal. Calcd: C18H11ClBrNO: C, 58.03; H, 2.93; N, 3.72. Found: C, 58.07; H, 4.95; N, 3.76.
3-(2-chlroquinolin-3-yl)-1-(3-nitrophenyl)-prop-2-en-1-one (3h)
Prepared by the preceding method from 1 (0.01 mol) and 3-nitroacetophenone (0.01 mol). Pale yellow solid. Yield: 80%, R f=0.54 (EtOAc/hexane, 4:6) MP: 145–150°C. MS (M+): 338.06 (100%), 340.04 (32%); FTIR (cm−1): 1732 (C=O), 1645 (CH=CH), 853 (C–Cl), 1589(–NO2); 1H NMR (400 MHz, DMSO-d6) Δ/ppm: 7.53 (1H, d, Hα), 8.51 (1H, d, Hβ), 7.43–8.10 (m, 1H, 2H, 3H, 4H, 5H, 1′H, 3′H, 4′H, 5′H). Anal. Calcd: C18H11ClN2O3: C, 68.71; H, 3.26; N, 8.24. Found: C, 63.84; H, 3.28; N, 8.28.
3-(2-chloroquinolin-3-yl)-1-(3, 4, 5-trimethoxyphenyl)-prop-2-en-1-one (3i)
Prepared by the preceding method from 1 (0.01 mol) and 3, 4.5-trimethoxyacetophenone (0.01 mol). Yellow crystalline solid. Yield: 87%. R f=0.48 (EtOAc/hexane, 3:7). MP: 174–175°C. MS (M+): 383.14 (100%), 385.09 (34%). FTIR (cm−1): 1732 (C=O), 1645 (CH=CH), 853 (C–Cl). 1H NMR (400 MHz, DMSO-d6) Δ/ppm: 7.53 (1H, d, Hα), 8.57 (1H, d, Hβ), 3.80 (s, 6H, CH3), 3.64 (s, 3H, CH3), 7.43–8.28 (m, 1H, 2H, 3H, 4H, 5H, 1′H, 5′H). Anal. Calcd: C21H18ClNO4: C, 65.71; H, 4.71; N, 3.64. Found: C, 65.84; H, 4.78; N, 3.68.
3-(2-chloroquinolin-3-yl)-1-(5-bromo-2-methoxyphenyl)-prop-2-en-1-one (3j)
Prepared by the preceding method from 1 (0.01 mol) and 2-methoxy-5-bromoacetophenone (0.01 mol). Yellow solid. Yield: 81%. R f=0.63 (EtOAc/hexane, 4:6). MP: 160–168°C. MS (M+): 400.87 (100%), 402.95 (75%). FTIR (cm−1): 1736 (C=O), 1645 (CH=CH), 853 (C–Cl), 588 (C–Br). 1H NMR (400 MHz, DMSO-d6) δ/ppm: 7.53 (1H, d, Hα), 8.57 (1H, d, Hβ), 3.73 (s, 3H, CH3), 7.40–8.31 (m, 1H, 2H, 3H, 4H, 5H, 2′H, 3′H, 5′H). Anal. Calcd: C20H14ClBrNO2: C, 56.61; H, 3.21; N, 3.44. Found: C, 56.64; H, 3.25; N, 3.46.
Conclusions
We have developed a K2CO3–Al2O3-catalyzed, simple, solvent-free, cost-effective, and environmentally benign technique for the synthesis of 3-(2-chloroquinolin-3-yl)-1-substituted phenyl prop-2-en-1-ones. These compounds have been synthesized in high yield by using K2CO3–Al2O3 and avoiding the use of any solvent under ultrasonicator. The attractive features of this procedure are the mild reaction conditions, high conversions, cleaner reaction profiles, solvent-free reaction conditions, operational simplicity, safety, environmental friendliness, and the inexpensive and readily available catalyst, which make the process greener.
Acknowledgements
We are thankful to the head of the Department of Chemistry, Rashtrasant Tukadoji Maharaj Nagpur University, Nagpur (India), for providing necessary laboratory facilities and to the director of SAIF Lucknow (India) for providing necessary spectral data of compounds.
References
- Nem , N.H. ; Kim , Y. ; You , Y.J. ; Hong , D.H. ; Kim , H.M. ; Ahn , B.Z. Eur. J. Med. Chem . 2003 , 38 , 179 – 187 .
- Wu , J.H. ; Wang , X.H. ; Yi , Y.H. ; Lee , K.H. Bioorg. Med. Chem. Lett . 2003 , 13 , 1813 – 1815 .
- Wu , X. ; Wilairat , P. ; Go , M.L. Bioorg. Med. Chem. Lett . 2002 , 12 , 2299 – 2302 .
- Tuchinda , P. ; Reutrakul , V. ; Claison , P. ; Pongprayoon , V. ; Sematong , T. ; Santisuk , A.T. ; Taylor , W.C. Phytochemistry 2002 , 59 , 169 – 173 .
- Xia , Y. ; Yang , Z.Y. ; Xia , P. ; Bastow , K.F. ; Nakanishi , Y. ; Lee , K.H Bioorg. Med. Chem. Lett . 2000 , 10 , 699 – 701 .
- Liu , M. ; Wilairat , P. ; Go , M.L. J. Med. Chem . 2001 , 44 , 4443 – 4452 .
- Rojas , J. ; Domínguez , J.N. ; Charris , J.E. ; Lobo , G. ; Paya , M. ; Ferrándiz , M.L. Eur. J. Med. Chem . 2002 , 37 , 699 – 699 .
- Fuentes , A. ; Marinas , J.M. ; Sinisterra , J.V. Tetrahedron Lett . 1987 , 28 , 4541 – 4544 .
- Climent , M.J. ; Corma , A. ; Iborra , S. ; Primo , J. J. Catal . 1995 , 151 , 60 – 77 .
- Ballini , R. ; Bosica , G. ; Ricciutelli , M. ; Maggi , R. ; Sartori , G. ; Sartorio , R. ; Righi , P. Green Chem . 2001 , 3 , 178 – 180 .
- Sebti , S. ; Solhy , A. ; Tahir , R. ; Boulaajaj , S. ; Mayoral , J.A. ; Fraile , J. ; Kossir , A. ; Ouminoun , H. Tetrahedron Lett . 2001 , 42 , 7953 – 7956 .
- Sebti , S. ; Solhy , A. ; Tahir , R. ; Simahi , A. Appl. Catal . 2002 , 235 , 273 – 278 .
- Saravanamurugan , S. ; Palanichamy , M. ; Arabindoo , B. ; Murugesan , V. J. Mol. Catal. A: Chem . 2004 , 218 , 101 – 111 .
- Climent , M.J. ; Corma , A. ; Iborra , S. ; Velty , A. J. Catal . 2004 , 221 , 474 – 482 .
- Mason , T.J. In Chemistry with Ultrasound ; Mason , T.J. ; Elsevier : London , 1990 ; pp. 65 – 133 .
- Lopez-Pestana , J.M. ; Avila-Rey , M.J. ; Martin-Aranda , R.M. Green Chem . 2002 , 4 , 628 – 630 .
- Berlan , J. ; Mason , T.J. Ultrasonics 1992 , 30 , 203 – 212
- Calvino-Casilda , V. ; López-Peinado , A.J. ; Martín-Aranda , R.M. ; Ferrera-Escudero , S. ; Durán-Valle , C.J. Carbon . 2004 , 42 , 1363 – 1369
- Sebti , S. ; Solhy , A. ; Smahi , A. ; Kossir , A. ; Oumimoun , H. Catal. Commun . 2002 , 3 , 335 – 337 .
- Perozo-Rondón , E. ; Martín-Aranda , R.M. ; Casal , B. ; Durán-Valle , C.J. ; Lau , W.N. ; Zhang , X.F. ; Yeung , K.L. Catal. Today 2006 , 114 ( 2–3 ), 183 – 187 .
- Thirunarayanan , G. ; Vanangamudi , G. Arkivoc 2006 , 12 , 58 – 64 .
- Jhala , Y.S. ; Dulawat , S.S. ; Verma , B.L. Indian J. Chem . 2006 , 45B , 466 – 469 .
- http://www.patientsville.com/toxic/sodium-hydroxide.htm .
- Wei Deng , Y. Xu , Q.X. Guo , 2005 , 16 ( 3 ), 327 – 330 .
- Gilman , H. ; Cason , L.F. J. Am. Chem. Soc . 1950 , 72 , 3468 – 3469 .
- Guttire , J.L. ; Rabjohn , N. J. Org. Chem . 1957 , 22 , 166 – 176 .
- Rochus , W. ; Kickuth , R. Chem. Abstr . 1961 , 56 , 10976 – 10986 .
- Calloway , N.O. ; Green , L.D. J. Am. Chem. Soc . 1937 , 59 , 809 – 883 .