Abstract
Monomode microwave-assisted syntheses of d-glucuronic and d-galacturonic acid derivatives are reported in the presence of a solid acid catalyst, consisting of sulfuric acid loaded onto silica. This approach affords a variety of surface-active monoglycosylated glucofuranosidurono-6,3-lactones and disubstituted galacturonic adducts in excellent yields in less than 10 min at 85°C. This study illustrates the application of microwave heating mode, in combination with a cost-effective solid catalyst, as an efficient, selective, and eco-friendly methodology in carbohydrate chemistry.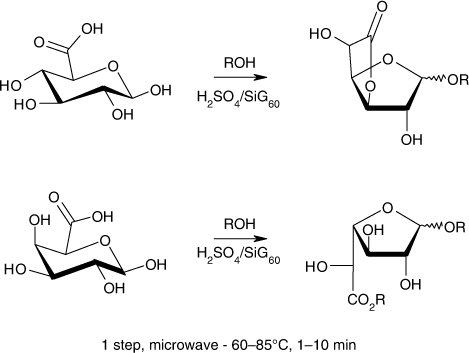
1. Introduction
In the context of the valorization of carbohydrates arising from agricultural resources (i.e. lignocellulosic feedstocks) Citation1, the quest of structurally homogeneous amphiphilic materials is one of the most developed research directions Citation2 Citation3. Among these carbohydrate-based biodegradable amphiphilic molecules, derivatives of d-glucuronic (d-GlcA) and d-galacturonic (d-GalA) acids, with a single or two alkyl chains, are efficient, biocompatible and bioresorbable, nonionic surface-active reagents Citation4–11. A panel of other synthetical uronic acid-containing entities find also special relevance in the design of bioactive molecules Citation12–16 as in supramolecular and materials chemistry Citation17–23.
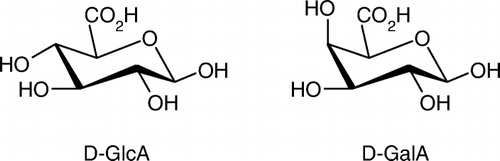
d-GlcA and d-GalA are naturally occurring hexuronic acids that are derived from renewable raw materials such as hemicellulose (i.e. glucurono-arabinogalactans) and pectins Citation24 Citation25. Containing two appropriate functional sites (anomeric hydroxyl and carboxylic acid) (), these monosaccharides are suited starting material for reactions with nucleophilic agents (alcohols, amines or thiols) Citation26. However, due to the same reactivity of both functional sites, the regioselective production of well-defined uronic acid derivatives, without generation of side-products, requires systematic multi-step pathways involving suitable protection, activation, and deprotection sequences Citation27 Citation28. In this context, the development of new sustainable, one-step and efficient strategies for the synthesis of anomerically pure uronic acid-based products is a particularly challenging task Citation10, Citation29, Citation30.
Scheme 1. Structures of β-d-glucuronic acid (d-GlcA) and β-d-galacturonic acid (D-GalA).
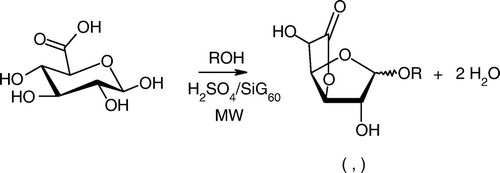
Previously, we disclosed that the tandem use of microwaves (MW) and heterogeneous catalysts could offer a convenient opportunity for the direct formation of uronic acid derivatives Citation31. Particularly, we reported on the application of sulfuric acid impregnated onto silica gel (H2SO4/SiG60) to convert quantitatively d-GlcA into monosubstituted β-d-glucofuranosidurono-6,3-lactones in a single step (). Reactions in closed vessels proceeded, in high yields and selectivities, after a few minutes (1–10 min) of microwave heating at 85°C with a maximum power of 80 Watt, which contrasted with previous works involving homogeneous Lewis acid conditions and extended reaction times Citation7, Citation10, Citation30. Moreover, it was notable that our solvent-free protocol presented attractive practical features, as easy set-up and convenient catalyst recovery (by filtration or centrifugation). The catalyst could also be reused for several consecutive batch reactions if carefully dried under static air at 160°C. Faced to environmental concerns, we anticipate herein that this approach may constitute a promising and low waste route to a range of new original tensioactive products.
Scheme 2. Microwave-promoted synthesis of monosubstituted lactones from glucuronic acid and alcohol (ROH).
As a part of our ongoing program dedicated to the elaboration of new chemical methodologies for the formation of high added-value materials from carbohydrates Citation31 Citation32, we evaluate the applicability of H2SO4/SiG60 to promote the “one-pot” modification of unprotected acidic sugars under microwave conditions.
2. Results and discussion
2.1. Microwave-assisted reaction between d-GlcA and various nucleophilic agents
Microwave-induced transformations were typically performed between 60 and 85°C, using a CEM Discover reactor, with a maximum power of 80 Watt under closed vessel conditions. Real-time temperature within the reaction vial was monitored using an infrared detector. In a first set of trial, d-GlcA was coupled with fivefold excess of alcohol in the presence of 35 wt% H2SO4/SiG60 (). We observed that, whatever the nature of the alcohol (ROH) investigated, d-GlcA conversions were nearly quantitative. After filtration of the solid catalyst through a glass frit and elimination of the excess of alcohol, the corresponding monosubstituted d-glucofuranosidurono-6,3-lactones 1-16 were recovered in good yields, mainly as the β anomer. No traces of disubstituted adducts (adducts where both the anomeric and the acid functions have been functionalized) and/or d-glucuronic acid γ-lactone were detected after microwave exposure, conversely to results obtained under classical thermally driven conditions Citation31. Structures of isolated products and anomeric configurations were unambigously assigned by NMR (Nuclear Magnetic Resonance) spectroscopy. These monosubstituted d-glucofuranosidurono-6,3-lactones are suited candidates for applications as surface-active agents Citation33.
Table 1. Microwave-assisted reaction between totally unprotected D-glucuronic acid and various nucleophilic agents in the presence of 35 wt% H2SO4/SiG60.a
Microwave-promoted reactions between d-glucuronic acid and alkyl alcohols afforded in almost quantitative yields the corresponding alkyl d-glucofuranosidurono-6,3-lactones 1–6 with high anomeric selectivities (α:β = 10:90) Citation31. Likewise, derivatisation of d-glucuronic acid with unsaturated moieties (such as allyl, butenyl, hexenyl, and undecenyl groups), in order to generate a new set of potent surface-active-agents, was conveniently achieved using our microwave strategy. Vinyl chains were selectively appended to the anomeric position in less than 10 min and without the recourse to solvent. Final lactonic products 7–10 were characterized by a typical Footnote1H NMR chemical shift around δ 5.04 ppm, associated to a lowfield resonance for the anomeric carbon at δ 109 ppm, clearly confirming the formation of the β-adduct. Incorporation of the vinyl fragment onto the sugar structure was also corroborated by 13C NMR signals around 135 and 115 ppm. Monosubstituted α-d-glucofuranosidurono-6,3-lactone was recovered as a minor by-product.
The feasibility of our microwave-assisted procedure was also underlined for the functionalization of d-GlcA with other representative alcohols: chloropropanol, benzyl alcohol, phenol, 1,6-hexanediol, and propargyl alcohol. In most cases, syntheses were conveniently achieved, providing in good yields the corresponding β-adducts 11–15 as the dominant forms. Noteworthingly, glycosylation between two sugar units and/or formation of diglycosylated adducts (for reaction with 1,6-hexanediol) were undetected.
Derivatization of the starting d-GlcA with phenol was not successful, mainly because of the fast oxidation of phenol under reaction conditions. The same conclusion was formulated when using p-cresol.
Finally, glycosylation of polyethylene glycol afforded quantitatively, after 10 min in the microwave reactor, the corresponding poly(ethylene glycol)-d-glucofuranosidurono-6,3-lactone 16 (α:β = 15:85). This reaction proceeded very quickly in less than 2 min at 85°C and enough for the quantitative conversion of d-GlcA. On the other hand, we demonstrated that the initial amount of PEG200 could noticeably be reduced to two equivalents without affecting both overall yield and anomeric selectivity of the reaction. However, work with an equimolar sugar/PEG ratio was unrealizable as decomposition of d-GlcA, by browning reaction, was observed.
Yields for compounds 7, 12, and 15 were lower than for other products, logically because of the lower reactivity of allyl, benzyl, and propargyl alcohols. This impacts also over the anomeric selectivity as a 25/75 α/β ratio was recorded for these three final lactones. Independently from reactivity concerns, the functionalization of d-GlcA with benzyl alcohol or PEG200 is particularly noteworthy. Indeed, only for these two specific alcohols, we observed experimentally a decomposition of the sample by browning reaction if extended reaction times (up to >10 min) and/or higher temperatures were applied. As previously mentioned by Gabriel et al. Citation34, the kinematic viscosity and the loss tangent value (tan δ) are two relevant parameters for microwave applications.1 For linear alkyl alcohols, as butanol, hexanol or octanol, the kinematic viscosity is ranging between 2.3 and 7.7 cP (at 25°C), while the loss tangent value decrease when increasing chain length (tan δ = 0.571, 0.344, and 0.181, respectively, for butanol, hexanol, and octanol). For benzyl alcohol and PEG200, these viscosity values attain, respectively, 8.0 cP and 50.0 cP and are associated to tan δ values of, respectively, 0.667 and >1.0. These high tan δ mean that both solvents are able to convert very efficiently microwave energy into heat energy and to transfer it quickly to reagents, leading probably to d-GlcA charring under viscous conditions.
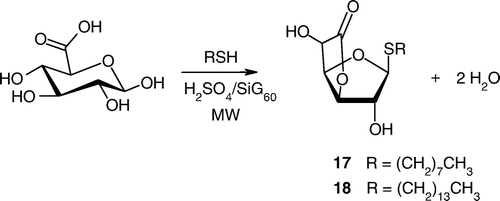
Finally, our approach was also extended to the functionalization of d-GlcA by alkyl thiols. We notably demonstrated that less than 5 min at 60°C was enough to recover compounds 17 and 18 in, respectively, 97 and 68% yields, exclusively as β-anomer ().
Scheme 3. Microwave-mediated functionalization of glucuronic acid by alkyl thiols (RSH).
These reactions are in agreement with the guidelines of green chemistry, as the application of microwave as an alternative heating source leads to improved yields and unprecedented selectivities. Moreover, our solvent-free protocol has a low environmental impact as water is the only side-product generated and as the catalyst can easily be recovered.
2.2. Microwave-assisted functionalization of mixtures of d-GlcA and d-glucose
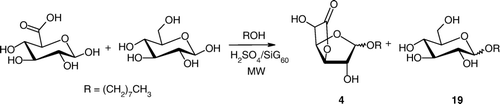
In order to substantially reduce both economical and environmental impacts related to the separation and the purification of sugars after hydrolysis of raw lignocellulosic materials, we proposed that our microwave strategy could also be exploited for the production of mixtures of surfactants. Thus, we considered the simultaneous benchmark derivatisation of d-glucose and d-glucuronic acid with n-octanol (). We anticipated that a synergy between both tensioactive molecules could be highlighted Citation35, Citation36. An equimolar mixture of both sugars and catalytic amounts of 35 wt% H2SO4/SiG60 in an fivefold excess of n-octanol was exposed to microwaves for 10 min at 85°C. NMR analysis of the crude reaction mixture, recovered after filtration of the solid acid promoter and elimination of the excess of alcohol, indicated the presence of octyl d-glucofuranosidurono-6,3-lactone 4 (98% yield based on d-glucose conversion, α:β = 5:95) and octyl d-glucopyranoside 19 (89% yield, α:β = 30:70). This result is quite interesting as it illustrates how silica sulfuric acid, in combination with microwaves, was able to catalyze efficiently and independently the alkylation of both sugars in high yields. As a fact, no evidence of a direct attachment between both sugars was underlined using two-dimensional NMR techniques.
Scheme 4. Simultaneous derivatisation of glucuronic acid and glucose.
2.3. Extension of the microwave-assisted protocol to d-galacturonic acid
The microwave-assisted protocol developed previously was applied to d-galacturonic acid (). Reactions performed under microwave exposure provided the corresponding functionalized galactosiduronates, mainly as the kinetically favored furanosidic isomers (). In this case, yields reached up to 85% and contrasted with previous work, involving extended reaction times and recourse to vacuum techniques Citation37. Unequivocal evidence of the predominant formation of α,β-furanosidic forms was supported by 13C NMR analyses (in combination with homonuclear and heteronuclear correlation experiments) with signals for C-4 around 83–85 ppm, more deshielded than in α,β-pyranuronate compounds (δC-4 typically close to 69–71 ppm) Citation38, Citation39.
Scheme 5. Microwave-synthesis of disubstituted galacturonic acid derivatives.
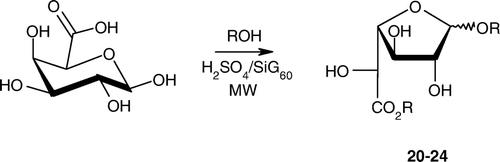
Table 2. Microwave-assisted reaction between totally unprotected D-galacturonic acid and various alcohols in the presence of 35 wt% H2SO4/SiG60.a
3. Experimental
d-glucuronic acid (≥98%), d-(+)-galacturonic acid monohydrate (≥97%), anhydrous d-(+)-glucose, and linear poly(ethylene glycol) (M n =200 g/mol) were purchased from commercial suppliers and used as received. Silica gel 60 was calcinated at 200°C prior to use. Solvents were dried using well established procedures. Thin-layer chromatography was performed on silica gel 60 plates (with fluorescent indicator F254) with visualization by charring with 10% H2SO4/ethanol solution. Preparative column chromatography was achieved using 230–400 mesh ASTM silica gel. 1H and 13C NMR, as two-dimensional spectra (gDQ-COSY, gHSQC-AD, gHMBC-AD, and NOESY), were recorded at 25°C on a Varian 600 MHz spectrometer. Chemical shifts are listed in parts per million downfield from TMS (tetramethylsilane) and are referenced by the residual solvent peaks. Infrared spectra were recorded at 4.0 cm−1 resolution on a Bruker IFS-48 FT-IR spectrometer. Melting points were estimated with a Büchi® melting point apparatus and were uncorrected. Microwave-assisted reactions were performed in specific 10 mL microwave reaction vials and irradiated in a CEM Discover® microwave reactor, instrument as KBr (potassium bromide) pellets. Mass spectra were acquired on an Esquire HCT high capacity ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with an electrospray ionization source. Best results were obtained in positive mode with capillary, nebulizer pressure, drying gas flow, and drying gas temperature set to 10,000 nA, 40.0 psi (nitrogen), 9.0 L min−1 (nitrogen), and 365°C, respectively. The scan range was adjusted to m/z 100–500 and the target mass set to 300. MS-data were recorded and processed using Bruker Daltonics data analysis software, version 3.0. All the mass spectra were acquired in ESI (electrospray ionization) in the positive mode.
3.1. Preparation of H2SO4/SiG60
The silica-supported sulfuric acid catalyst was prepared according to a well-established procedure Citation40 by incipient wetness impregnation of silica gel 60 (with BET specific surface area of 500±50 m2/g and average pore diameter of 60±5 Å) with an appropriated aqueous solution of H2SO4 yielding catalyst with an acid loading of 35 wt% (i.e. 35 g pure H2SO4/100 g final catalyst).
3.2. General procedure for the microwave-assisted synthesis and purification of compounds 1–24
Carbohydrates (d-GlcA, d-GalA, and d-glucose), alcohol/thiol (5 equiv.), and the acid catalyst were introduced together in a specific 10 mL pressure-resistant closed microwave vial. This vial was next placed within the microwave monomode cavity and irradiated under stirring. Control of the temperature during the experiment was assigned by an infrared detector. After heating, reactions were immediately quenched by forced compressed air cooling of the vial. Crude mixtures were filtrated to eliminate solid acid catalyst, washed with CHCl3, and then classically chromatographied on silica gel. When possible, the alpha anomeric form was also isolated for at least NMR analysis.
Spectral and physico-chemical data for alkyl α,β-d-glucofuranosidurono-6,3-lactones 1–7 Citation31 Citation41–43, n-octyl glucoside 19 Citation44, methyl (methyl α,β-d-galactofuranosid)uronate 20 Citation43 Citation45 Citation46, butyl (butyl α,β-d-galactosid)uronate 21, hexyl (hexyl α,β-d-galactosid)uronate 22, and octyl (octyl α,β-d-galactosid)uronate 24 (37) were in agreement with those reported in the literature.
3.2.1. Allyl d-glucofuranosidurono-6,3-lactone 7
Mixture of α and β isomers: colorless oil. Only the β anomer was isolated. 7β: off-white solid (recristall ised in light petroleum); mp 104–106°C. IR (KBr) ν 3440 cm−1, 3017 cm−1, 2976 cm−1, 1758 cm−1. 1H NMR (600 MHz, D2O) δ 5.72 (m, 1H, OCH2CH=CH2), 5.23 (m, 2H, OCH2CH = CH 2), 5.08 (s, 1H, H-1), 4.92 (dd, 1H, J 3,4 4.8 Hz, J 4,5 6.0 Hz, H-4), 4.59 (d, 1H, J 3,4 4.8 Hz, H-3), 4.26 (s, 1H, H-2), 4.10 (m, 1H, OCH 2CH = CH2), 3.96 (d, 1H, J 4,5 6.0 Hz, H-5), 3.87 (m, 1H, OCH2CH = CH2). 13C NMR (D2O) δ 177.28 (C = O), 136.53 (CH = CH2), 115.43 (CH = CH2), 107.43 (C-1), 83.32 (C-3), 77.87 (C-4), 76.24 (C-2), 68.79 (C-5), 62.53 (OCH2). ESI-HRMS+ calculated for C9H12LiO6 [M + Li]+ m/z 223.1306, found 223.1204.
3.2.2. 3′-butenyl d-glucofuranosidurono-6,3-lactone 8
Mixture of α and β isomers: colorless oil. α and β isomers were isolated. IR (NaCl) ν 3429 cm−1, 2933 cm−1, 1783 cm−1. 8β: 1H NMR (600 MHz, CDCl3) δ 5.77 (m, 1H, CH=CH2), 5.08 (m, 2H, CH = CH 2), 5.05 (s, 1H, H-1), 4.96 (dd, 1H, J 3,4 3.6 Hz, J 4,5 6.6 Hz, H-4), 4.81 (d, 1H, J 3,4 3.6 Hz, H-3), 4.35 (d, 1H, J 4,5 6.6 Hz, H-5), 4.33 (s, 1H, H-2), 3.72 (m, 2H, OCH2), 2.30–2.25 (m, 2H, CH2). 13C NMR (CDCl3) δ 174.69 (C = O), 134.77 (CH = CH2), 117.49 (CH = CH2), 109.05 (C-1), 83.18 (C-3), 77.31 (C-2), 77.04 (C-4), 69.17 (C-5), 68.08 (OCH2), 33.47 (CH2CH = CH2). 8α: 13C NMR (CDCl3) δ 174.69 (C = O), 102.48 (C-1), 84.72 (C-3), 76.82 (C-2), 76.02 (C-4), 70.07 (C-5), 69.12 (OCH2), 33.78 (CH2CH = CH2).
3.2.3. 5′-hexenyl d-glucofuranosidurono-6,3-lactone 9.
Mixture of α and β isomers: colorless oil. Only the β anomer was isolated. 9β: IR (NaCl) ν 3418 cm−1, 2937 cm−1, 2864 cm−1, 1785 cm−1. 1H NMR (600 MHz, CDCl3) δ 5.74 (m, 1H, CH=CH2), 5.02 (s, 1H, H-1), 4.94 (m, 3H, H-4, CH = CH 2), 4.81 (d, 1H, J 3,4 4.2 Hz, H-3), 4.38 (d, 1H, J 4,5 6.6 Hz, H-5), 4.29 (s, 1H, H-2), 3.66 (m, 1H, OCH2), 3.33 (m, 1H, OCH2), 2.04-1.98 (q, 2H, J 6.6 Hz, CH 2CH = CH2), 1.54-1.48 (m, 2H, OCH2 CH2), 1.38 (m, 2H, CH2). 13C NMR (CDCl3) δ 175.13 (C = O), 138.56 (CH = CH2), 114.55 (CH = CH2), 109.03 (C-1), 83.25 (C-3), 77.28 (C-2), 77.07 (C-4), 69.18 (C-5), 68.53 (OCH2), 33.32 (CH2CH = CH2), 31.90, 28.49, 24.94. ESI-HRMS+ calculated for C12H19LiO6 [M + Li]+ m/z 265.2199, found 265.2204.
3.2.4. 10′-undecenyl d-glucofuranosidurono-6,3-lactone 10
Mixture of α and β isomers: off-white solid. Only the β anomer was isolated. 10β: white solid (recristallised in Et2O). mp 77–79°C. IR (KBr) ν 3440 cm−1, 2924 cm−1, 2854 cm−1, 1758 cm−1. 1H NMR (600 MHz, CDCl3) δ 5.78 (m, 1H, CH=CH2), 5.04 (s, 1H, H-1), 4.96 (m, 2H, H-4, CH = CH 2), 4.89 (d, 1H, CH = CH2), 4.83 (d, 1H, J 3,4 4.2 Hz, H-3), 4.36 (d, 1H, J 4,5 7.2 Hz, H-5), 4.33 (s, 1H, H-2), 3.59 (m, 1H, OCH2), 3.37 (m, 1H, OCH2), 2.02-1.99 (q, 2H, J 6.6 Hz, CH 2CH = CH2), 1.54-1.48 (m, 2H, OCH 2CH2), 1.25 (m, 12H, CH2). 13C NMR (CDCl3) δ 174.69 (C = O), 139.17 (CH = CH2), 114.08 (CH = CH2), 109.08 (C-1), 83.18 (C-3), 77.32 (C-2), 77.03 (C-4), 69.14 (C-5, OCH2), 33.76 (CH2CH = CH2), 29.51, 29.45, 29.16, 28.89, 25.68.
3.2.5. 3′-Chloropropyl d-glucofuranosidurono-6,3-lactone 11
Mixture of α and β isomers: yellow oil. Only the β anomer was isolated. 11β: colorless oil. TLC (EtOAc/n-hexane 1/1 v/v): R f 0.17. IR (NaCl) ν 3345 cm-1, 2964 cm-1, 2888 cm-1, 1783 cm-1, 1052 cm-1. 1H NMR (600 MHz, CDCl3) δ 5.06 (s, 1H, H-1), 4.98 (dd, 1H, J 3,4 3.6 Hz, J 4,5 6.6 Hz, H-4), 4.84 (d, 1H, J 3,4 3.6 Hz), 4.41 (d, 1H, J 4,5 6.6 Hz, H-5), 4.34 (s, 1H, H-2), 3.85 (m, 2H, OCH2), 3.47 (t, 2H, J 7.2 Hz, CH2Cl), 1.94 (m, 2H, OCH2CH 2). 13C NMR (CDCl3) δ 175.07 (C = O), 109.19 (C-1), 83.17 (C-3), 77.47 (C-2), 76.85 (C-4), 69.32 (C-5), 65.03 (OCH2), 41.76 (CH2Cl), 31.94 (OCH2 CH2).
3.2.6. Benzyl d-glucofuranosidurono-6,3-lactone 12
Mixture of α and β isomers: yellow oil. IR (NaCl) ν 3395 cm−1, 3034 cm−1, 2879 cm−1, 1793 cm−1. Only the β anomer was isolated. 12β: TLC (EtOAc/n-hexane 1/1 v/v): R f 0.12. 1H NMR (600 MHz, CDCl3) δ 7.41–7.33 (m, 5H, Harom), 5.07 (s, 1H, H-1), 4.79 (dd, 1H, J 3,4 3.6 Hz, J 4,5 6.6 Hz, H-4), 4.62 (m, 3H, CH 2Ph, H-3), 4.29 (s, 1H, H-2), 4.16 (d, 1H, J 4,5 6.6 Hz, H-5). 13C NMR (CDCl3) δ 174.97 (C = O), 140.84 (Cispo), 128.45 (Cmeta), 128.30 (Cpara), 127.56 (Cortho), 108.51 (C-1), 83.14 (C-3), 77.61 (C-2), 76.92 (C-4), 70.41 (C-5), 66.81 (OCH2). ESI-HRMS+ calculated for C13H14LiO6 [M + Li]+ m/z 273.1929, found 273.1932.
3.2.7. Cyclohexyl d-glucofuranosidurono-6,3-lactone 13
Mixture of α and β anomers: colorless oil. Only the β anomer was isolated. 13β: IR (NaCl) ν 3416 cm−1, 2946 cm−1, 2862 cm−1, 1786 cm−1. 1H NMR (600 MHz, CDCl3) δ 5.21 (s, 1H, H-1), 4.95 (dd, 1H, J 3,4 4.8 Hz, J 4,5 6.6 Hz, H-4), 4.82 (d, 1H, J 3,4 4.8 Hz, H-3), 4.33-4.31 (m, 2H, H-5, H-2), 3.74-3.68 (m, 1H, OCH), 1.85 (m, 2H, CH2), 1.69 (m, 2H, CH2), 1.51 (d, 1H, J 6.0 Hz, CH2), 1.28-1.12 (m, 5H, CH2). 13C NMR (CDCl3) δ 174.39 (C = O), 106.79 (C-1), 83.23 (C-3), 77.84 (C-2), 76.81 (C-4), 70.35 (OCH), 68.99 (C-5), 32.71 (CH2), 25.40 (CH2), 24.09 (CH2). ESI-HRMS+ calculated for C12H18LiO6 [M + Li]+ m/z 265.2169, found 265.2164.
3.2.8. 6′-hydroxy-n-hexyl d-glucofuranosidurono-6,3-lactone 14
Mixture of α and β isomers: yellow oil. Only the β anomer was isolated. 14β: IR (NaCl) ν 3345 cm−1, 2932 cm−1, 2859 cm−1, 1782 cm−1. 1H NMR (600 MHz, CDCl3) δ 5.06 (s, 1H, H-1), 4.98 (dd, 1H, J 3,4 4.8 Hz, J 4,5 7.2 Hz, H-4), 4.82 (d, 1H, J 3,4 4.8 Hz, H-3), 4.35 (m, 2H, H-5, H-2), 3.71 (m, 2H, OCH2), 3.61 (m, 2H, HOCH2), 1.52 (OCH2CH 2), 1.33 (CH2). 13C NMR (CDCl3) δ 175.92 (C = O), 109.45 (C-1), 83.33 (C-3), 77.46 (C-2), 77.12 (C-4), 69.48 (C-5), 62.80 (OCH2), 62.18 (OCH2), 32.48 (OCH2CH2), 25.71 (CH2).
3.2.9. Propargyl d-glucofuranosidurono-6,3-lactone 15
Mixture of α and β isomers: colorless oil. IR (NaCl) ν 3440 cm−1, 2938 cm−1, 2875 cm−1, 2121 cm−1, 1783 cm−1. α and β isomers were isolated. 15β: 1H NMR (600 MHz, DMSO-d 6) δ 5.09 (s, 1H, H-1), 4.77 (dd, 1H, J 3,4 4.8 Hz, J 4,5 6.6 Hz, H-4), 4.70 (d, 1H, J 3,4 4.8 Hz, H-3), 4.41 (d, 1H, J 4,5 6.6 Hz, H-5), 4.21 (s, 2H, OCH2), 4.06 (s, 1H, H-2), 3.35 (s, C≡CH). 13C NMR (DMSO-d 6) δ 175.27 (C = O), 106.55 (C-1), 84.42 (C≡CH), 83.01 (C-3), 78.82 (C-4), 77.49 (C-2), 75.15 (C≡CH), 69.02 (C-5), 55.83 (OCH2). 15α: 13C NMR (DMSO-d 6) δ 175.27 (C = O), 101.62 (C-1), 85.00 (C-3), 84.41 (C≡CH), 79.75 (C-2), 76.82 (C-4), 76.17 (C≡CH), 69.09 (C-5), 55.04 (OCH2).
3.2.10. Poly(ethylene glycol) d-glucofuranosidurono-6,3-lactone 16
Mixture of α and β isomers: light yellow oil. Only the β anomer was isolated. 16β: IR (NaCl) ν 3382 cm−1, 2877 cm−1, 1789 cm−1. 1H NMR (600 MHz, D2O) δ 5.07 (s, 1H, H-1), 4.93 (m, 2H, H-3, H-4), 4.59 (d, 1H, J 5,4 6.0 Hz, H-5), 4.28 (s, 1H, H-2), 3.72 (m, 1H, OCH2), 3.58 (m, 1H, OCH2), 3.45 (br, CH2-PEG). 13C NMR (D2O) δ 177.28 (C = O), 108.37 (C-1), 83.40 (C-3), 77.74 (C-4), 76.18 (C-2), 68.95 (C-5), 66.99 (OCH2).
3.2.11. n-Octanethio-d-glucofuranosidurono-6,3-lactone 17
Isolated as the β anomer. Light yellow oil. 1H NMR (600 MHz, CDCl3) δ 5.17 (s, 1H, H-1), 4.44 (dd, 1H, H-5), 4.32 (d, 1H, H-4), 3.74 (t apparent, 1H, H-3), 3.42 (d, 1H, H-2), 2.62 (m, 2H, SCH2), 1.54 (m, 2H, SCH2CH 2), 1.26 (m, 10H, CH2), 0.81 (t, 3H, CH3). 13C NMR (CDCl3) δ 173.8 (C = O), 90.5 (C-1), 86.16 (C-4), 70.68 (C-5), 54.9 (C-2), 48.1 (C-3), 31.7 (SCH2), 29.8, 29.4, 29.3, 28.9, 28.8, 24.5 (CH2), 13.9 (CH3).
3.2.12. n-Tetradecanethio-d-glucofuranosidurono-6,3-lactone 18
Isolated as the β anomer. Light brown oil. 1H NMR (600 MHz, CD3OD) δ 4.99 (s, 1H, H-1), 4.86 (dd, 1H, J 4,5 6.5 Hz, H-4), 4.79 (d, 1H, J 3,4 4.1 Hz, H-3), 4.51 (d, 1H, J 5,4 6.5 Hz, H-5), 4.16 (s, 1H, H-2), 3.78 (dt, 1H, -OCH2), 3.26 (dt, 1H, -OCH2), 1.48 (m, 2H, -OCH2 CH2), 1.33-1.22 (m, 22H, (CH2)11), 0.88 (t, 3H, CH3). RMN 13C (CD3OD) : δ (ppm) = 175.6 (C-6), 109.06 (C-1), 84.3 (C-3), 78.1 (C-2), 77.8 (C-4), 70.8 (C-5), 67.83 (-OCH2), 31.64, 29.36, 29.32, 29.28, 29.23, 29.16, 29.03, 28.83, 25.70, 22.29, (-CH2), 13.1 (CH3).
3.2.13. PEG (PEG d-galactosid)uronate 24
Mixture of furanosid and pyranosid forms. PEG (PEG β-d-galactofuranosid)uronate 21β: yellow oil. TLC (EtOAc): R f 0.12. IR (NaCl) ν 3410 cm−1, 1750 cm−1. 13C NMR (D2O) δ 172.88 (C = O), 107.47 (C-1), 83.33 (C-4), 80.66 (C-2), 75.71 (C-3), 69.47 (PEG), 68.27 (C-5), 66.86 (OCH2), 64.72 (CO2CH2), 60.39 (PEG).
4. Conclusion
In summary, we have illustrated that combination of microwave and a supported acid catalyst can be used to carry out the one-step functionalization of two representative acidic carbohydrates in short times and in solventless conditions. This straightforward approach can prove beneficial as the solid catalyst was removed by filtration at the end of the process, thus minimizing wastes. The advantage of microwaves is evident as high yields were associated to high chemo- and anomeric-selectivities.
Acknowledgements
This work was carried out in the framework of the “Technose” Excellence Program supported by the “Région Wallonne” (Belgium). The authors are grateful to the “Région Wallonne” for its financial support.
Notes
1. The loss factor (tan δ) is defined as the ratio between dielectric loss (ϵ″) and the dielectric constant (ϵ′) and is used to quantify the ability of a material to convert microwaves into heat energy at a given temperature and frequency. For practical applications, at 2.45 GHz and 20°C, solvents could be categorized into “excellent” (with tan δ > 0.6–0.5), “medium” (0.5 > tan δ > 0.1) or “poor” (tan δ < 0.1) candidates for microwave applications.
References
- Lichtenthaler , F.W. In : Methods and Reagents for Green Chemistry ; Tundo , P. , Perosa , A. , Zecchini , F. , Wiley : Hoboken , 2007 , pp 23 – 63 .
- Burczykn , B. In Novel Surfactants ; Holmberg , K. ; Surfactant Science Series ; Marcel Dekker : New York , 2003 ; Vol. 114 , pp 129 – 192 .
- de Almeida , M.V. ; Le Hyaric , M. Mini-Rev. Org. Chem . 2005 , 2 3 , 283 – 297 .
- Blecker , C. ; Danthine , S. ; Pétré , M. ; Lognay , G. ; Moreau , B. ; Vander Elst , L. ; Paquot , M. ; Deroanne , C. J. Coll. Interf. Sci . 2002 , 321 1 , 154 – 158 .
- Moreau , B. ; Lognay , G. ; Blecker , C. ; Destain , J. ; Gerbaux , P. ; Chéry , F. ; Rollin , P. ; Paquot , M. ; Marlier , M. Biotechnol. Agron. Soc. Environ . 2007 , 11 , 9 – 17
- Blecker , C. ; Piccicuto , S. ; Lognay , G. ; Deroanne , C. ; Marlier , M. ; Paquot , M. J. Coll. Interf. Sci . 2008 , 247 , 424 – 428 .
- Bertho , J.-N. ; Ferrières , V. ; Plusquellec , D. J. Chem. Soc. Chem. Commun . 1995 , 1391 – 1393 .
- Auvray , X. ; Labulle , B. ; Petipas , C. ; Bertho , J.-N. ; Benvegnu , T. ; Plusquellec , D. J. Mater. Chem . 1997 , 7 8 , 1373 – 1376 .
- Bernard , D. EP Patent 0819,698 A2 , 1997 .
- Ferrières , V. ; Bertho , J.-N. ; Plusquellec , D. Carbohydr. Res . 1998 , 311 1 – 2 , 25 – 35 .
- Ferlin , N. ; Grassi , D. ; Ojeda , C. ; Castro , M.J.L. ; Grand , E. ; Fernández Cirelli , A. ; Kovensky , J. Carbohydr. Res . 2008 , 343 , 839 – 847 .
- Mignet , N. ; Chaix , C. ; Rayner , B. ; Imbach , J.-L. Carbohydr. Res . 1997 , 303 , 17 – 24 .
- Stachulski , A.V. ; Jenkins , G.V. Nat. Prod. Rep . 1998 , 15 , 173 – 186 .
- Kim , Y.J. ; Ichikawa , M. ; Ichikawa , Y. J. Org. Chem . 2000 , 65 8 , 2599 – 2602 .
- Röckendorf , N. ; Lindhorst , T.K. J. Org. Chem . 2004 , 69 , 4441 – 4445 .
- Doyle , D. ; Murphy , P.V. Carbohydr. Res . 2008 , 343 , 2535 – 2544 .
- Hashimoto , K. Polym. Appl . 1999 , 48 10 , 460 – 468 .
- Haupt , M. ; Knaus , S. ; Rohr , T. ; Gruber , H. J. Macromol. Sci. Part A 2000 , 37 4 , 323 – 341 .
- Shimura , Y. ; Hashimoto , K. ; Yamanaka , C. ; Setojima , D. J. Polym. Sci. Part A: Polym. Chem . 2001 , 39 22 , 3893 – 3903 .
- Mahrholz , T. ; Klein , J. ; Klein , T. Macromol. Chem. Phys . 2002 , 203 18 , 2640 – 2649 .
- Weiss , S.L. ; Sieverling , N. ; Niclasen , M. ; Maucksch , C. ; Thünemann , A.F. ; Möhwald , H. ; Reinhardt , D. ; Rosenecker , J. ; Rudolph , C. Biomaterials 2006 , 27 , 2302 – 2312 .
- Bergman , K. ; Hilborn , J. ; Bowden , T. Express Polym. Lett . 2008 , 2 8 , 553 – 559 .
- Doyle , D. ; Murphy , P. Carbohydr. Res . 2008 , 343 , 2535 – 2544 .
- Sun , R. ; Lawther , J.M. ; Banks , W.B. Carbonhydr. Polym . 1996 , 29 , 325 – 331 .
- Jar vis , M.C. ; Forsyth , W. ; Duncan , H.J. Plant Physiol . 1988 , 88 , 309 – 314 .
- Timoshchuk , V.A. Russ. Chem. Rev . 1995 , 64 , 675 – 703 .
- Pilgrim , W. ; Murphy , P.V. Org. Lett . 2009 , 11 4 , 932 – 942 .
- Tampure , R.P. ; Strecker , T.E. ; Chaplin , D.J. ; Siim , B.G. ; Trawick , M.L. ; Pinney , K.G. J. Nat. Prod . 2010 , 73 6 , 1093 – 1101 .
- El Alaoui , A. ; Schmidt , F. ; Monneret , C. ; Florent , J.C. J. Org. Chem . 2006 , 71 26 , 9628 – 9636 .
- Rat , S. ; Mathiron , D. ; Michaud , P. ; Kovensky , J. ; Wadouachi , A. Tetrahedron 2007 , 63 , 12424 – 12428 .
- Richel , A. ; Laurent , P. ; Wathelet , B. ; Wathelet , J.-P. ; Paquot , M. Tetrahedron Lett . 2010 , 51 , 1356 – 1360 .
- Richel , A. ; Laurent , P. ; Wathelet , B. ; Wathelet , J.-P. ; Paquot , M. Catal. Today 2011 , 167 , 141 – 147 .
- Razafindralambo , H. ; Blecker , C. ; Richel , A. ; Laurent , P. ; Richard , G. ; Wathelet , B. ; Wathelet , J.P. ; Paquot , M. Abstract Book of the 5th Edition of World Congress on Emulsion . 2010 , p 98 .
- Gabriel , C. ; Gabriel , S. ; Grant , E.H. ; Halstead , B.S.J. ; Mingos , D.M.P. Chem. Soc. Rev . 1998 , 27 , 213 – 223 .
- Garamus , V.M. ; Milkerei , G. ; Gerber , S. Vill , V. Chem. Phys. Lett . 2004 , 392 , 105 – 109 .
- Golemanov , K. ; Denkov , N.D. ; Tcholakova , S. ; Vethmathu , M. ; Lips , A. Langmuir 2008 , 24 , 9956 – 9961 .
- Petit , S. EP Patent 0532,370 B1 , 1998 .
- Ritchie , R.G.S. ; Cyr , N. ; Korsch , B. ; Koch , H.J. ; Perlin , A.S. Can. J. Chem . 1975 , 53 , 1424 – 1433 .
- Ramos , M.L.D. ; Caldeira , M.M.M. ; Gil , V.M.S. Carbohydr. Res . 1996 , 286 , 1 – 15 .
- Kogelbauer , A. ; Vassena , D. ; Prins , R. ; Armor , J.N. Catal. Today 2000 , 55 , 151 – 160 .
- Cadotte , J.E. ; Smith , F. ; Spriestersbach , D. J. Am. Chem. Soc . 1952 , 74 , 1501 – 1504 .
- Madaj , J. ; Trynda , A. ; Konitz , A. ; Nowacki , A. ; Wisniewski , A. Carbohydr. Res . 1998 , 308 , 431 – 433 .
- Bordoni , A. ; de Lederkremer , R.M. ; Marino , C. Tetrahedron 2008 , 64 , 1703 – 1710 .
- Kurashima , K. ; Fujii , M. ; Ida , Y. ; Akita , H. Chem. Pharm. Bull . 2004 , 52 2 , 270 – 275 .
- Matsuhiro , B. ; Zanlungo , A.B. ; Dutton , G.G.S. Carbohydr. Res . 1981 , 97 , 11 – 18 .
- Bordoni , A. ; Lima , C. ; Mariño , K. ; de Lederkremer , R.M. ; Marino , C. Carbohydr. Res . 2008 , 343 , 1863 – 1869 .