Abstract
2,10-Dichloro-12-trichloromethyl-6-substituted xanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin-6-sulfides (2a–l) with the eight-membered phosphorus and oxygen heterocyclic ring were synthesized by reacting equimolar quantities of alcohols, carbon disulfide, and 2,6,10-trichloromethyl-12H-dibenzo[d,g][1,3,2] dioxaphosphocin-6-sulfide Citation1 in the presence of dimethylaminopyridine (DMAP). The structures of the products were confirmed by IR, 1H, 13C, 31P-NMR, and mass spectral analyses.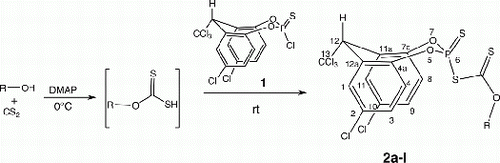
Introduction
The dibenzophosphocins acquired much importance recently due to their insecticidal, bacterial, and anticarcinogenic properties Citation1 . Organophosphonates with heteroatoms in the α/β-positions are gaining attention because of their interesting biological properties Citation2–5.
They are found to inhibit rennin, HIV proteases Citation6 Citation7 and catalytic antibodies Citation8. Particularly their thiophosphoryl xanthates exhibited potential fungicidal and herbicidal Citation9 properties. Due to growing concern for the toxic influence of organic solvents on the environment and the living system, organic synthesis without solvents has been the primary object of organic chemists Citation10. In view of this, solvent-free synthesis of new class of dibenzodioxaphosphocin-6-sulfide with a xanthate group without solvent has been accomplished.
Synthesis and discussion
The synthetic route () involves the preparation of title compounds (2a–l) in a one pot solvent-free reaction in the presence of 4-dimethylaminopyridine (4-DMAP). Equimolar quantities of alcohols and carbon disulfide are reacted at 0 °C to form the xanthate complex intermediate which on further reaction with dibenzodioxaphosphocin monochloride (1) at 0 °C afforded 2a–l in high yields Citation11. When the same reaction was carried out in the presence of other bases like triethylamine, diisopropyl ethylamine, or 1,8-diazabicyclo [5,4,0] undec-7-ene, the yields of these compounds are very poor. However, with 4-DMAP as catalyst, the reaction were completed even at 0 °C affording excellent yields of the products. Further, a variety of sterically hindered and functionalized substrates also reacts under similar reaction conditions successfully affording high product yields. These compounds separated from the reaction mixture as brown colored gummy semi-solids and they were purified by column chromatography on silica gel (gradient elution with 0–20% ethyl acetate in hexane).
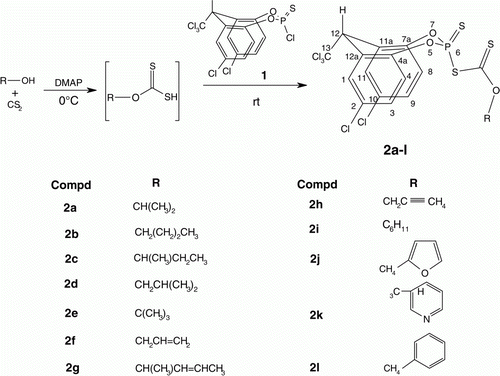
The IR spectra of 2a–l showed characteristic absorption bands at 1262–1237 (C–S) Citation12, 1240–1175 (C=S) Citation13, 761–741 (P=S) Citation14 Citation15, and 660–625 cm−1(P–S) Citation12. In the 1H-NMR spectra, the six aromatic protons of the dibenzodioxaphosphocin moiety in all compounds (2a–l) showed only three sets of signals, each signal accounting for two protons, because of their symmetrical disposition in dibenzodioxaphosphocin ring Citation16. The doublets at δ 8.08–8.14 (d, J=1.7–2.4 Hz), δ 7.19–7.75 (dd, J=1.7–2.5, 8.4–9.1 Hz), and δ 6.97–7.26 (d, J=6.5–8.7 Hz) are assigned to 1 & 11-H, 3 & 9-H, and 4 & 8-H, respectively. The bridged 12-H methylene protons appeared as doublet at δ 6.02–6.06 (d, J=1.5 Hz) due to its long range coupling (5 J PH) with phosphorous Citation11. The dibenzophosphocin system in all these compounds 2a–l appeared to exist in rigid boat-chair conformation with both cis and trans configurations Citation17 and the 5 J PH coupling “through space” appeared to occur since the electrons of the ring oxygen atoms and the C-12 proton are on the same side of the heterocyclic ring Citation17. Protons of 6-alkyl/alkenyl/benzyl moieties of the title compounds 2a–l gave signals in the expected range of δ 0.85–8.41.
13C-NMR spectra of 2a–l showed expected peaks at their expected positions. Oxygen bearing C(4a) and C(7a) resonated as a doublet in the region δ 145.6–147.2 (2 J POC(4a,7a)=6.0–7.1 Hz). The doublet at δ 124.8–125.6 (3 J POCC(4,8)=4.2–4.7 Hz) was assigned to C(4) and C(8) Citation17. The chemical shift of bridged C(11a) and C(12a) appeared as singlets at δ 132.0–132.8 Citation11. The chlorine substituted C(2) and C(10) gave signals at δ 131.0–132.3. Chemical shifts at δ 127.6–128.9 and 129.4–130.8 were assigned to C(3) & C(9) and C(1) & C(11), respectively. The C(12) carbon signal appeared as a singlet in the down field region at δ 52.7–53.2 Citation18 due to the deshielding effect of trichloromethyl group and C(13) carbon chemical shift appeared at δ 97.5–98.1 Citation15. The C(2′) chemical shift of the xanthato moiety appeared downfield in all the compounds when compared to the corresponding carbon chemical shift in the respective free alcohols Citation13. All the compounds except 2g and 2l exhibit two 31P-NMR chemical shifts, in the range of δ 41.0–59.8 and this may be attributed to the presence of two conformers for them in the solution state.
Experimental
The melting points were determined in open capillary tubes on a Mel-Temp apparatus and are uncorrected. IR spectra (νmax, cm−1) were recorded in KBr pellets on Perkin-Elmer 1000 unit. The 1H, 13C, 31P-NMR, and Mass spectra were recorded on Varian Gemini 300 and Varian AM 400 MHz NMR spectrometer operating at 300 and 400 MHz for 1H, 75.46 and 100.57 MHz for 13C, and 121.7 MHz for 31P. All the compounds were dissolved in CDCl3 and chemical shifts were referred to those of TMS (1H and 13C) and 85% H3PO4 (31P). Microanalytical data were obtained from Central Drug Research Institution, Lucknow, India.
General procedure for the synthesis of 2,10-dichloro-12-tricholoromethyl-6-n-isopropylxanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2a)
Isopropanol (0.01 mol) and 4-DMAP (0.005 mol) mixture was stirred for 5 min at room temperature (rt) and cooled to 0 °C. Carbon disulfide (0.005 mol) was added to it and further stirred for 20 min. 2,6,10-tricholoro-12-tricholoromethyl-6-butylxanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (1, 0.005 mol) Citation10 was added to it and the reaction mixture was kept stirred at rt for 30 min till the TLC analysis indicated completion of the reaction. The gummy syrup separated was purified over silica gel column chromatography using ethylacetate: hexane (1:3) as an eluent. Other compounds 2b-l were synthesized by adopting the above procedure.
(2a): Yield 2.60 g (86%), mp 172–174 °C. IR (KBr) cm−1: 1262 (C–S), 1235 (C=S), 761 (P=S), 635 (P–S); 1H-NMR (CDCl3): δ 8.13 (d, J=2.2 Hz, 2H, 1 & 11-H), 7.75 (dd, J =2.3,8.9 Hz, 2H, 3 & 9-H), 7.26 (d, J=8.7 Hz, 4 & 8-H), 6.06 (d, J=1.5 Hz, 1H, 12-H), 4.80–4.69 (m, 1H, OCH), 1.24 (d, J=10.2 Hz, 6H, CH3); 13C-NMR (CDCl3) δ 172 (C=S), 129.7 (C-1 & 11), 131.3 (C-2 & 10), 128.7 (C-3 & 9), 125.0 (C-4 & 8), 146.0 (d, J=6.0 Hz, C-4a & 7a), 132.0 (C-11a & 12a), 53.1 (C-12), 98.0 (C-13), 60.0 (d, J=5.5 Hz,-OCH), 22.0 (s, CH3); 31P (CDCl3): 52.0, 54.0 ppm; GC-MS m/z (%): 579 (50, M+.), 581 (18, M + 2). Anal. Calcd for C18H14Cl5O3PS3: C, 37.10; H, 2.42. Found: C, 37.01; H 2.30.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-n-butylxanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2b)
Yield 2.75 g (89%), mp 166–168 °C. IR (KBr) cm−1: 1261 (C–S), 1214 (C=S), 745 (P=S), 635 (P–S); 1H-NMR (CDCl3): δ 8.12 (d, J=2.4 Hz, 2H, 1 & 11-H), 7.19 (dd, J=2.4,9.0 Hz, 2H, 3 & 9-H), 6.97 (d, J=8.7 Hz, 2H, 4 & 8-H), 6.05 (d, J=1.5 Hz, 1H, 12-H), 4.41–4.34 (m, 2H, OCH2), 1.82–1.75 (m, 2H, CH2). 1.51–1.44 (m, 2H, CH2), 0.96–0.85 (t, J=10.8 Hz, 3H, CH3); 13C-NMR (CDCl3) δ 173 (C=S), 129.4 (C–1 & 11), 131.0 (C-2 & 10), 127.8 (C-3 & 9), 125.2 (d, J=1.8 Hz, C-4 & 8), 145.6 (d, J=6.8 Hz, C-4a & 7a), 132.0 (C-11a & 12a), 53.1 (C-12), 97.5 (C-13), 69.1 (d, J=5.5 Hz, -OCH2), 31.0 (d, J=7.5 Hz, -OCH2-CH2), 17.7 (d, J=13.2 Hz, -OCH2-CH2-CH2), 12.6 (s, CH3); 31P (CDCl3): 48.5, 53.7 ppm; GC-MS m/z (%): 593 (65, M+.), 595 (21, M + 2). Anal. Calcd for C19H16Cl5O3PS3: C 38.24, H 2.70. Found: C 38.12, H 2.61.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-sec-butylxanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2c)
Yield 2.59 g (84%), mp 178–179 °C. IR (KBr) cm−1: 1260 (C–S), 1214 (C=S), 741 (P=S), 632 (P–S); 1H-NMR (CDCl3): δ 8.12 (d, J=1.7 Hz, 2H 1 & 11-H), 7.62 (dd, J=2.2, 8.6 Hz, 2H, 3 & 9-H), 7.20 (d, J=6.7 Hz, 2H, 4 & 8-H), 6.04 (d, J=1.4 Hz, 1H, 12-H), 3.68–5.58 (m, 1H, OCH), 1.76–1.73 (m, 2H, CH2), 1.12 (d, J=10.2 Hz, 3H, CH-CH3), 0.99 (t, J=10.5 Hz, 3H, CH3); 13C-NMR (CDCl3): δ 171 (C=S), 129.9 (C-1 & 11), 131.2 (C-2 & 10), 128.9 (C-3 & 9), 125.1 (d, J=1.8 Hz, C-4 & 8), 146.2 (d, J=6.2 Hz, C-4a & 7a), 132.2 (C-11a & 12a), 53.08 (C-12), 97.8 (C-13), 72.4 (d, J=6.8 Hz, -OCH), 32.2 (-OCH-CH2), 20.2 (-OCH-CH3), 13.6 (s, -CH2-CH3); 31P (CDCl3): 45.2, 51.1 ppm Anal. Calcd for C19H16Cl5O3PS3: C 38.24, H 2.70. Found: C 38.10, H 2.78.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-iso-butylxanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2d)
Yield 2.81 g (91%), mp 184–185 °C. IR (KBr) cm−1: 1263 (C–S), 1240 (C=S), 742 (P=S), 626 (P–S); 1H-NMR (CDCl3): δ 8.11 (d, J=1.8 Hz, 2H, 1 & 11-H), 7.40 (dd, J=1.7, 8.8 Hz, 2H, 3 & 9-H), 7.22 (d, J=8.0 Hz, 2H, 4 & 8-H), 6.04 (d, J=1.6 Hz, 12-H), 4.30 (d, J=7.2 Hz, 2H, OCH2), 2.25–2.11 (m, 1H, CH), 1.04 (d, J=10.2 Hz, 6H, CH3); 13C-NMR (CDCl3): δ 172 (C=S), 129.8 (C-1 & 11), 131.4 (C-2 & 10), 128.2 (C-3 & 9), 125.0 (d, J=1.8 Hz, C-4 & 8), 146.4 (d, J=6.0 Hz, C-4a & 7a), 132.4 (C-11a & 12a), 53.1 (C-12), 97.9 (C-13), 67.2 (d, J=5.8 Hz, -OCH2-CH), 29.5 (d, J=7.8 Hz, -OCH2-CH), 19.1 (-CH-CH3); 31P (CDCl3): 41.0, 43.0 ppm Anal. Calcd for C19H16Cl5O3PS3: C 38.24, H 2.70. Found: C 38.14, H 2.62.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-tert-butylxanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2e)
Yield 2.50 g (81%), mp 176–177 °C. IR (KBr) cm−1: 1237 (C–S), 1175 (C=S), 741 (P=S), 625 (P–S); 1H-NMR(CDCl3): δ 8.14 (d, J=2.2 Hz, 2H, 1 & 11-H), 7.30 (d, J=2.3 Hz, 2H, 3 & 9-H), 7.19 (d, J=7.2 Hz, 2H, 4 & 8-H), 6.04 (d, J=1.4 Hz, 1H, 12-H), 1.33 (s, 9H, CH3); 13C-NMR (CDCl3): δ 173 (C=S), 130.0 (C-1 & 11), 131.2 (C-2 & 10), 128.0 (C-3 & 9), 125.1 (d, J=1.8 Hz, C-4 & 8), 146.0 (d, J=6.8 Hz, C-4a & 7a), 132.7 (C-11a & 12a), 52.7 (C-12), 98.0 (C-13), 68.4 (-OC-CH3), 31.3 (-OCCH3); 31P (CDCl3): 45.0, 48.2 ppm. Anal. Calcd for C19H16Cl5O3PS3: C 38.24, H 2.70. Found: C 38.10, H 2.63.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-allyl xanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2f)
Yield 2.45 g (82%), mp 168–169 °C. IR (KBr) cm−1: 1262 (C–S), 1235 (C=S), 745 (P=S), 660 (P–S); 1H-NMR(CDCl3): δ 8.10 (d, J=2.2 Hz, 2H, 1 & 11-H), 7.42 (dd, J=2.3, 9.0 Hz, 2H, 3 & 9-H), 7.20 (d, J=7.4 Hz, 2H, 4 & 8-H), 6.03 (d, J=1.4 Hz, 1H, 12-H), 6.09–5.96 (m, 1H, CH), 5.43–5.37 (m, 2H, CH2). 4.03 (d, J=4.5 Hz, 2H, CH2); 13C-NMR (CDCl3): δ 172 (C=S), 130.8 (C-1 & 11), 132.2 (C-2 & 10), 127.8 (C-3 & 9), 125.2 (C-4 & 8), 146.0 (d, J=6.8 Hz, C-4a & 7a), 132.8 (C-11a & 12a), 53.0 (C-12), 98.1(C-13), 64.0 (d, J=7.2 Hz, -OCH2-CH),137.0 (-OCH-CH2) 115.0 (-CH-CH2); 31P (CDCl3): 54.0, 59.8.; GC-MS m/z (%): 577 (45, (M+?), 579 (14, M + 2). Anal. Calcd for C18H12Cl5O3PS3: C 37.23, H 2.08. Found: C 37.11, H 2.01.
Synthesis of 2,10-dichlor-12-tricholoromethyl-6-methyl propenyl carbonyl xanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2g)
Yield 2.59 g (82%), mp 225–227 °C. IR (KBr) cm−1: 1262 (C–S), 1240 (C=S), 748 (P=S), 637 (P–S); 1H-NMR (CDCl3): δ 8.08 (d, J=2.2 Hz, 2H, 1 & 11-H), 7.30 (dd, J=2.3, 8.7 Hz, 2H, 3 & 9-H), 7.20 (d, J=6.9 Hz, 2H, 4 & 8-H), 6.02 (d, J=1.3 Hz, 12-H), 5.62–5.45 (m, 2H, CH), 4.40–4.20 (m, 1H, CH),1.90 (d, J=10.2 Hz, 3H, CH3), 1.20 (d, J=10.5 Hz, 3H, CH3); 13C-NMR (CDCl3): δ 172 (C=S), 130.4 (C-1 & 11), 132..0 (C-2 & 10), 127.9 (C-3 & 9), 125.6 (d, J=1.8 Hz, C-4 & 8), 147.2 (d, J=6.8 Hz, C-4a & 7a), 132.6 (C1a & 12a), 53.1 (C-12), 98.0 (C-13), 70.0 (-OCH), 25.2 (-OCH-CH3), 136.0 (-CH=CH-CH3), 128.0 (CH=CH-CH3), 18.2 (CH=CH-CH3); 31P (CDCl3): 45.0 ppm; GC-MS m/z (%): 605 (39, M+.), 607 (11, M + 2). Anal. Calcd for C20H16Cl5O3PS3: C 39.46, H 2.65. Found: C 39.32, H 2.56.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-propargyl xanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2h)
Yield 2.38 g (80%), mp 148–150 °C. IR (KBr) cm−1: 1262 (C–S), 1235 (C=S), 761 (P=S), 632 (P–S); 1H-NMR(CDCl3): δ 8.12 (d, J=1.7 Hz, 2H, 1 & 11-H), 7.42 (dd, J=1.7, 8.4 Hz, 2H, 3 & 9-H), 7.11 (d, J=8.1 Hz, 2H, 4 & 8-H), 6.03 (d, J=1.4 Hz, 1H, 12-H), 4.30–4.26 (m, 1H), 1.86–1.14 (s, 1H); 13C-NMR (CDCl3) δ 174 (C=S), 130.0 (C-1 & 11), 132.1 (s, C-2 & 10), 127.6 (C-3 & 9), 125.4 (C-4 & 8), 146.8 (d, J=6.2 Hz, C-4a & 7a), 132.4 (s, C-11a & 12a), 53.0 (C-12), 97.8 (C-13), 57.5 (CH2=C), 78.8 (CH2=C), 72.0 (CH2-C-CH); 31P (CDCl3): 41.0, 44.0 ppm. Anal. Calcd for C18H10Cl5O3PS3: C 37.36, H 1.74 Found: C 37.21, H 1.69.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-cyclohexyl xanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2i)
Yield 2.69 g (83%), mp 170–172 °C. IR (KBr) cm−1: 1261 (C–S), 1215 (C=S), 750 (P=S), 626 (P–S); 1H-NMR (CDCl3): δ 8.13(d, J=1.8 Hz, 2H, 1 & 11-H), 7.38 (dd, J=1.6,9.1 Hz, 2H, 3 & 9-H), 7.21 (d, J=8.7 Hz, 2H, 4 & 8-H), 6.03 (d, J=1.4 Hz, 1H, 12-H), 4.30–4.26 (m, 1H, Cyclohexyl), 1.86–1.14 (m, 10H, Cyclohexyl); 13C-NMR (CDCl3): δ 175 (C=S), 130.2 (C-1 & 11), 132.0 (C-2 & 10), 127.7 (C-3 & 9), 125.4 (C-4 & 8), 146.8 (d, J=6.2 Hz, C-4a & 7a), 132.4 (C-11a & 12a), 53.0 (C-12), 97.8 (C-13), 77.5 (C-1′), 31.8 (C-2′ & C-6′) 22.0 (C-3′ & C-5′) 26.8 (C-4′); 31P (CDCl3): 45.0, 57.2 ppm. Anal. Calcd for C21H18Cl5O3PS3: C 40.50, H 2.91. Found: C 40.40, H 2.80.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-furfuryl xanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2j)
Yield 2.71 g (84%), mp 194–195 °C. IR (KBr) cm−1: 1262 (C–S), 1214 (C=S), 740 (P=S), 635 (P–S); 1H-NMR (CDCl3): δ 8.12 (d, J=1.9 Hz, 2H, 1 & 11-H), 7.30 (dd, J=2.0, 8.9 Hz, 2H, 3 & 9-H), 7.21 (d, J=6.9 Hz, 2H, 4 & 8-H), 6.03 (d, J=1.4 Hz, 1H, 12-H), 7.41–7.38 (d, J=4.4 Hz, 1H, Ar–H), 6.42–6.36 (m, 2H, Ar–H), 4.62 (s, 2H, OCH2); 13C-NMR (CDCl3): δ 173 (C=S), 130.8 (C-1 & 11), 132.3 (C-2 & 10), 128.0 (C-3 & 9), 125.0 (d, J=4.2 Hz, C-4 & 8), 146.3 (d, J=7.0 Hz, C-4a & 7a), 132.6 (C-11a & 12a), 53.2 (C-12), 97.6 (C-13), 58.0 (OCH2), 152.0 (C-2′), 108.0 (C-3′), 111.0 (C-4′), 144.0 (C-5′); 31P (CDCl3): 50.4, 53.2 ppm. Anal. Calcd for C20H12Cl5O4PS3: C 38.70, H 1.95. Found: C 38.60, H 1.90.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-nicotinyl xanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2k)
Yield 2.89 g (88%), mp 204–205 °C. IR (KBr) cm−1: 1263 (C–S), 1240 (C=S), 741 (P=S), 660 (P–S); 1H-NMR (CDCl3): δ 7.18 (d, J=6.5 Hz, 2H, 4 & 8-H), 6.05 (d, J=1.5 Hz, 1H, 12-H), 8.41–7.33 (m, 8H), 4.62–4.58 (s, 2H, OCH2); 13C-NMR (CDCl3): δ 173 (C=S), 130.0 (C-1 & 11), 132.0 (C-2 & 10), 128.2 (C-3 & 9), 124.8 (C-4 & 8), 146.8 (d, J=6.2 Hz, C-4a & 7a), 132.4 (C-11a & 12a), 53.1 (C-12), 97.6 (C-13), 62.0 (OCH2), 152.0 (C-2′), 130.0 (C-3′), 135.2 (C-4′), 125.0 (C-5′), 145.2 (C-6′); 31P (CDCl3): 52.0, 55.2 ppm. Anal. Calcd for C21H13Cl5NO3PS3: C 39.92, H 2.07, N 2.22. Found: C 39.80, H 2.00, 2.14.
Synthesis of 2,10-dichloro-12-tricholoromethyl-6-benzyl xanthato-12H-dibenzo[d,g] [1,3,2] dioxaphosphocin 6-sulfide (2l)
Yield 2.82 g (86%), mp 210–211 °C. IR (KBr) cm−1: 1261 (C–S), 1238 (C=S), 743 (P=S), 642 (P–S); 1H-NMR (CDCl3): δ 8.12 (d, J=1.9 Hz, 2H, 1 & 11-H), 6.03 (12-H), 7.35–7.33 (m, 9H), 4.72 (s, 2H, OCH2); 13C-NMR (CDCl3): δ 173 (s, C=S), 130.5 (C-1 & 11), 132.0 (C-2 & 10), 128.6 (C-3 & 9), 125.0 (C-4 & 8), 145.9 (d, J=7.1 Hz, C-4a & 7a), 132.6 (C-11a & 12a), 53.2 (C-12), 98.0 (C-13), 65.2 (OCH2), 138.8 (C-1′), 126.8 (C-4′ & C-6), 129.4 (C-3′ & C-5′), 128.0 (C-6′); 31P (CDCl3): 54.0 ppm. Anal. Calcd for C22H14Cl5O3PS3: C 41.89, H 2.24. Found: C 41.80, H 2.20.
Conclusion
In summary, we have carried out a study on the synthesis of a series of new eight-membered phosphorus and oxygen heterocyclic containing xanthato phosphocin sulphide derivatives. A convenient and efficient approach was developed under mild conditions in good yields of products. The results obtained show that the study is significant to the synthetic organic chemistry.
Acknowledgements
The authors thank Prof. C. D. Reddy, Department of Chemistry, S.V. University, Tirupati for helpful discussions and CSIR, Human Resources Development Group, Government of India, New Delhi for providing financial assistance (01/2347/09/EMR-II).
References
- Ananda Kumar , K. ; Kuasturaiah , M. ; Suresh Reddy , C. ; Berlin , K.D. J. Heterocyclic Chem . 2003 , 40 , 345 – 351 .
- Engel , R. 1992 . Handbook of Organophosphorus Chemistry , New York : Marcel Dekker .
- Fest , C. and Schmidt , K.J. 1973 . The Chemistry of Organophosphorus Pesticides , Berlin , NY : Springer-Verlag .
- Paula , V.F. ; Barbosa , L.C.A. ; Teixeira , R.R. ; Picanço , M.C. ; Silva , G.A. Pest Manag. Sci . 2008 , 64 , 863 – 872 .
- Kiran , Y.B. ; Devendranath Reddy , C. ; Gunasekar , D. ; Naga Raju , C. ; Barbosa , L.C.A. ; Marney , D.C.O. ; Russell , L.J. J. Fire Sci . 2007 , 25 , 193 – 215 .
- Stowassor , B. ; Budt , K.-H. ; Jian-Qi , L. ; Peyman , A. ; Ruppert , D. Tetrahedron Lett . 1992 , 33 , 6625 – 6628 .
- Liu , X. ; Li , Z. ; MeKenna , C.E. Phosp. Sul. Sili . 1999 , 147 , 213 – 214 .
- Hirschman , R. ; Smith , A.B. III ; Taylor , C.A. ; Bencovic , P. ; Taylor , S.D. Yager , K.M. ; Sprengeler , P.A. ; Bencovic , S.J. Science . 1994 , 265 , 234 – 237 .
- Richardson , R.G. Weed Res . 1980 , 20 , 159 – 163 .
- Knochel , P. , Houk , K.N. , Kessler , H. , Modern Solvents in Organic Synthesis, Topic In Current Chemistry ; Berlin , NY : Springer-Verlag , 1999 ; vol 206 .
- Ananda Kumar , K. ; Kasturaiah , M. ; Suresh Reddy , C. ; Devendranath Reddy , C. Synth. Comm . 2003 , 33 , 819 – 825 .
- Mc Ivor , R.A. ; Grant , G.A. ; Hubley , C.E. Can J. Chem . 1956 , 34 , 1611 – 1640 .
- Clerc , P. and Simon , S. 1985 . Spectral Data of Structure Determination of Organic Compounds , 1235 Berlin , NY : Springer-Verlag .
- Ibrahime , E.H.M. , Alnaimi , I.S. and Nour , E.M. 1987 . Quatar Univ. Sci . Bull. , 7 : 81 – 90 .
- Chittenden , R.A. ; Thomas , L.C. Spectrochemica Acta . 1966 , 22 , 1449 – 1463 .
- Odorisio , P.A. ; Pastor , S.D. ; Spivack , J.D. ; Bini , D. ; Rodenbough , R.K. Phosp. Sul. Sili . 1984 , 19 , 285 – 293 .
- Goddard , J.D. ; Payne , A.W. ; Cook , N. J. Heterocyclic Chem . 1988 , 25 , 575 – 588 .
- Devendranath Reddy , C. ; Sankar Reddy , B. ; Mallikarjuna Reddy , P. ; Darrell Berlin , K. ; Couch , K.M. ; Tyagi , S. ; Hossain , M.B. ; Der Helm , D.K. Phosp. Sul. Sili . 1996 , 115 , 149 – 160 .