
ABSTRACT
Ultrasonic and microwave-assisted practical methods have been developed for the nitration of phenols using metal nitrates in aqueous polyethylene glycol (PEG) media. Solvent is recycled three times for reproducibility. It was recycled with minimum loss and decomposition. Developed protocols were cost effective, simple and efficient, which afforded nitration products in good to excellent yields. The observed hyperchromic/hypochromic shifts in the UV/VIS spectra of metal nitrates in PEG solutions could be due to the plausible in situ formation of “PEG-bound M(II) nitrate” and thereby the release of nitronium ion () during the course of reaction when metal nitrate [M(II)nitrate] is added to PEG. Nitronium thus formed most likely is captured by aromatic compound to afforded nitro aromatics.

Introduction
Aromatic compounds undergo electrophilic substitution due to the presence of high electron density delocalised on the benzene ring. Nitration reaction is an industrially important electrophilic substitution reaction for the synthesis of nitro-aromatic products, which are widely used as solvents, and intermediates in the manufacture of pharmaceuticals, synthetic dyestuffs and other fine chemicals (
Citation1–3). These reactions are difficult to proceed even under drastic conditions. The conventional nitration procedure involves the use of “acid mixture”, which produces nitronium ion (), an active species required for nitration. Acid mixture used in the direct nitration methods of aromatic compounds contains large amounts of nitric and sulphuric acids (HNO3 and H2SO4), which are highly toxic, corrosive, and pollutes the environment. A large number of protocols still use organic solvents as reaction media to facilitate the reaction, many of which are volatile, flammable, sometimes explosive, and have a damaging effect on human health and/or on the environment. Constant attention has been paid all over the world in the design and execution of mild methods of nitration of aromatic compounds to overcome these defects and prevent/minimise environmental pollution (
Citation4–10). On the advice of US Environmental Protection Agency, Paul Anastas and John Warner developed 12 principles of green and sustainable chemistry which include (a) the use of safe, environment-benign substances, including solvents, whenever possible; (b) the design of energy-efficient processes; (c) the design of processes which allow the use of maximum amount of raw material to convert into the product; and (d) the best form of waste disposal to protect safe environment in the first place (
Citation11). The use of a green solvent is also an important alternative solution for minimisation and protection of environmental pollution (
Citation11). Properties of ideal green solvent demand that it should be non-toxic, inexpensive, recyclable, capable to dissolve a great range of organic compounds and possess high boiling point, low vapour pressure. But there is a remote possibility to find such an effective green solvent. Perhaps water is only one of the greener solvents one can imagine because it is clean, non-toxic, inexpensive, the most environmentally benign and abundantly available in nature. It is well-known fact that several reactions that occur in living beings are carried out in water and are catalysed by enzymes (
Citation12). Prior to the 1980s, the use of water as a solvent was limited to hydrolysis reactions. Many striking examples have appeared in the literature showing that water has unique properties as a solvent that can lead to surprising results. The pioneering works of Breslow (
Citation13), Grieco (
Citation14), Lindström (
Citation15), and Corma and Garcia (
Citation16) provided excellent findings of water-mediated reactions and related bibliography. Despite the fact that it is the cheapest, safest and most non-toxic solvent in the world, its presence is generally avoided through the dehydrative drying of substrates and solvents. The use of water as a medium for organic reactions is therefore one of the latest challenges for modern organic chemists. Pioneering green chemistry principles of Anastas and Warner also became stimulus to design and perform organic reactions under solvent-free conditions which make synthesis simpler, save energy, and prevent solvent wastes, hazards, and toxicity (
Citation17, Citation18). Within no time these reactions received much attention because of their enhanced selectivity, mild reaction conditions, and associated ease of manipulation. The other side of energy and time-saving aspects of green chemical approach to organic synthesis is ultrasonic (
Citation19–22) and microwave- (
Citation23–26) assisted organic reactions. In recent years aqueous solutions of polyethylene glycol (PEG) have been widely used as solvents or catalysts in a different kind of reactions in organic synthesis (
Citation27) due to their important environmentally benign characteristics such as low-toxicity, low volatility, biodegradability and low cost as a bulk commodity chemical. In a review article Chen et al. (
Citation28) elaborately highlighted that PEG, and solutions of PEG have been successfully used as green reaction media in a variety of substitution, oxidation and reduction reactions (
Citation29). In addition, aqueous PEG solutions quite often have been used as good substitutes for expensive and toxic phase transfer catalysts (
Citation30). Encouraged by these aspects, in the present study, we successfully developed acid-free synthetic protocols for nitration of aromatic compounds by replacing conventional “acid mixture (mixture of HNO3and H2SO4)” in aqueous PEG media using a variety of metal nitrates under conventional and ultrasonic and microwave-assisted conditions.
Results and discussion
Nitric acid is one of the well-known oxidising agents even in dilute solutions. Dilute nitric acid in bulk water system can oxidise the chloride ion (Cl−) ion to Cl2 and Br− to Br2 if water contains salts in high concentrations. Nitration reactions underwent smoothly with metal nitrates in the presence of PEGs. Nitration of phenols was performed by metal nitrates (1.1 mol dm–3) in PEG systems at 90°C. The nitronium ion (), the active species for nitration or oxidation, can be generated not only in aqueous PEG systems but also in bulk water containing salts in high concentrations. Just as the water structure of nanoscale water droplets in reversed micelle systems is distorted, that of “bulk” aqueous solution can be also distorted by salts in high concentrations and, consequently, “bulk” aqueous solutions should lose the properties of the bulk water. In the reactions using Zn(NO3)2, which occur in a homogeneous medium, the ultrasound-mediated reactions have led to complete conversion in a very short period (40–45 min) with much higher para selectivity than those reported so far. Reaction rates are accelerated with the introduction of electron-donating groups and are retarded with electron-withdrawing groups.
Effect of structure on reactivity
To check the generality of the reaction, an array of substituted phenols and metal nitrates are used under varied reaction conditions, as shown in Scheme 1. In order to have a closer look into the effect of structural variation of phenol on nitration, the study has been taken up extensively the following variable (in solution phase) conditions
Different metal nitrates Zn(NO3)2, Cu(NO3)2, Ni(NO3)2, Zr(NO3)2, Cr(NO3)2, Cd(NO3)2, Co(NO3)2 and AgNO3.
Different polyethylene glycols (PEG-200, -300, -400, -600, -4000 and -6000).
Scheme 1. Nitration of phenols under different conditions.

Data presented in indicate that the PEG-triggered reactions are too sluggish even after 24 h, under conventional conditions irrespective of the metal nitrate used. Yet the reaction appeared to be sensitive to the structural variation of PEG. Among the several systems presented in , Zn(NO3)2-triggered nitration in PEG-400 has been found to be more effective than other PEGs. According to Pearson's theory (
Citation31) of “Hard and Soft Acids and Bases” a hard metal interacts with a base in much the same way as a proton, by binding to a lone pair of electrons on the base. The stability of complexes of hard acids with hard bases increases as the ligand becomes more basic. Furthermore, it is important to note that the metal nitrates used herein belong to the transition metal group. Because of the available vacant d-orbitals they can easily form “PEG-bound M(II) nitrate” adducts during the course of reaction. In order to have a closer look into this aspect, we have scanned UV/VIS spectra of certain metal nitrates (with and without PEG-400) under experimental conditions. The results are presented in . UV/VIS spectroscopic data presented in show that spectral bands of metal nitrates underwent either hypochromic or hyperchromic shift in the presence of PEG-400, indicating metal nitrates form “PEG-bound M(II)nitrate adducts” in situ during the course of chemical reaction. Thus, the mechanism of the reaction could be explained through in situ formation of “PEG-bound M(II) nitrate” and there by the release of nitronium ion (), during the course of reaction when metal nitrate [M(II)nitrate] is added to PEG. Nitronium thus formed most likely is captured by aromatic compound to afford nitro aromatics as shown in Scheme 2.
Scheme 2. Proposed mechanism for the nitration of aromatic compounds in the presence of PEG.
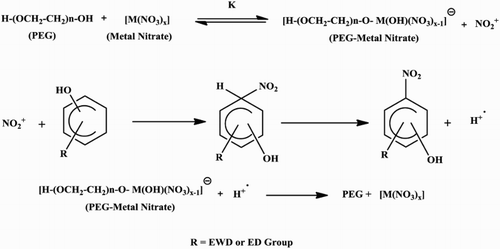
Table 1. Metal nitrate mediated nitration of phenol in the presence of PEG under conventional conditions.
Table 2. Interaction of PEG-400 with different metal nitrates.
Ultrasonic-assisted organic synthesis (USAOS) is a powerful and green approach which is being used to accelerate synthesis of organic compounds. It is an environmentally benign synthesis, which minimised the use of the precious metal catalysts and led to the development of new eco-friendly protocols ( Citation20–23). The chemical effects of ultrasound do not come from a direct interaction with molecular species but arises from acoustic cavitation; the formation, growth, and implosive collapse of bubbles in a liquid ( Citation20–23). The observed rate and yield enhancements observed in the present study () could be attributed to ultrasonic cavitation effect. Reaction times decreased enormously in ultrasonic and microwave-assisted protocols. The developed methods afforded good to excellent yields of products with high efficiency. After obtaining successful results in USAOS methods we were enthusiastic to see whether these reaction times could be further reduced under solvent-free conditions using microwave irradiation. The frequency used in heating applications is usually 2450 MHz (wavelength 12.2 cm). The energy of microwaves is so low that only molecular rotation could be induced. Microwaves have no effect on molecular bonds or electron clouds such as infrared (IR) or the visible region of electromagnetic radiation has. Results obtained under microwave-assisted synthesis (MWAS) are compiled in , which clearly indicate highly remarkable rate accelerations (reaction times reduced from several (≥20) to few minutes), followed by high yields. This dramatic rate enhancement could be attributed to bulk activation of molecules, which is believed to be due to rapid superheating of the polar solvents and pressure effects ( Citation24–26).
Table 3. Nitration of certain aromatic compounds (phenols) using Zn(NO3)2 in PEG-400.
Experimental details
Materials and methods: All chemicals purchased from Aldrich, Merck, Loba or Fluka were of reagent grade. Progress of the reactions was monitored by thin layer chromatography (TLC) and visualisation was accomplished by UV light or by I2. Products of the reactions were characterised by spectroscopic methods and physical data such as melting/boiling points. Melting points were recorded on BUCHI B-545 capillary melting point apparatus and are uncorrected. IR spectra were recorded on a Perkin Elmer Fourier transform infrared spectrometer. Nuclear magnetic resonance spectra were recorded at a Varian VNMRS-400 MHz spectrometer. Chemical shifts are reported as values in ppm relative to CHCl3 (7.26), and tetramethylsilane was used as internal standard. Mass spectra were recorded on a ZAB-HS mass spectrometer using spectroscopy, electrospray ionization mass spectrometry ionisation.
Conventional synthesis: In a typical reaction, phenol (1.0 mmol) was dissolved in 1 M PEGs solution (10 ml) and metalnitrate was added (1.1 mmol) and heated up to 90°C, the progress of the reaction was monitored by TLC. After the completion of reaction, the organic layer was diluted with ethylacetate (10 ml), and separated from the aqueous layer. It was then washed with (3 × 5 ml) water, separated and finally dried over sodium sulphate. The anhydrous ethylacetate layer was concentrated in reduced pressure to afford the crude product, which was subjected to column chromatography (silica gel, 100–200 mesh) using EtOAc–hexane (3:7) as eluent to obtain the pure product. Solvent is recycled three times for reproducibility. After isolation of the product from the reaction system, the mother liquor (the reaction mixture of PEG-600 and water) was extracted with ether because PEG is insoluble in ether. The ether layer was decanted, and mother liquor dried for 4 h under the IR light or the aqueous filtrate was distilled directly at 100°C to remove water. The recovered PEG-400 can be reused for consecutive runs. It was recycled with minimum loss and decomposition. The next run was performed using the same conditions.
Ultrasonically assisted synthesis: In a typical reaction, phenol (1.0 mmol) was dissolved in 1 M PEGs solution (10 ml) and metalnitrate was added (1.1 mmol) was sonicated at room temperature (27°C) in an ultrasonic cleaning bath. The ultrasonic bath had a frequency of 33 kHz and electric power rating of 100 W. The reaction was carried out in a round bottom flask of 50 ml capacity equipped with a mechanical agitator and the flask was suspended at the centre of the ultrasonic bath. The progress of the reaction was monitored by TLC. After the completion of reaction, the organic layer was diluted with ethylacetate (10 ml), and separated from aqueous layer. It was then washed with (3 × 5 ml) water, separated and finally dried over sodium sulphate. The anhydrous ethylacetate layer was concentrated in reduced pressure to afford the crude product, which was subjected to column chromatography (silica gel, 100–200 mesh) using EtOAc–hexane (3:7) as eluent to obtain the pure product.
MWAS: Phenol (1.0 mmol) was dissolved in 1 M PEGs solution (10 ml) and metalnitrate was added (1.1 mmol) in a microwave vessel and subjected into a microwave reactor and the progress of the reaction was monitored by TLC. After the completion of the reaction, the organic layer was diluted with ethylacetate (10 ml), and separated from the aqueous layer. It was then washed with (3 × 5 ml) water, separated and finally dried over sodiumsulphate. The anhydrous organic layer was concentrated in vacuum to afford the crude product, which was subjected to column chromatography (silica gel, 100–200 mesh) using EtOAc–hexane (3:7) as eluent to obtain the pure product.
Spectral data of certain nitro phenols
2-NO2 phenol: δ10.52 (s 1H, OH), 8.15 (dd 1H, J = 8.5 Hz, J = 8 Hz), 7.55 (dd 1H, J = 8 Hz, J = 7.5 Hz), 6.95 (d 1H, J = 8.5 Hz), 8.12 (d 1H, J = 8 Hz); m/z = 139.
4-NO2 phenol: δ10.95 (s 1H, OH), 6.95 (d 2H, J = 8 Hz), 8.15 (d 2H, J = 8 Hz); m/z = 139.
2-Me-4-NO2 phenol: δ10.55 (s 1H, OH), 2.35 (s 3H, Me), 6.85 (d 1H, J = 8.5 Hz), 8.12 (d 1H, J = 8.5 Hz), 8.25 (dd 1H, J = 8.5 Hz, J = 7.5 Hz); m/z = 153.
2-Me-6-NO2 phenol: δ10.25 (s 1H, OH), 2.35 (s 3H, Me), 7.44 (dd 1H, J = 8 Hz, J = 8 Hz), 7.26 (d 1H, J = 8 Hz), 8.15 (d 1H, J = 8 Hz); m/z = 153.
2-NO2-4-Me phenol: δ10.42 (s 1H, OH), 2.42 (s 3H, Me), 7.32 (d 1H, J = 8 Hz), 7.12 (d 1H, J = 8 Hz), 7.92 (s 1H); m/z = 153.
3-Me-4-NO2 phenol: δ10.64 (s 1H, OH), 2.35 (s 3H, Me), 6.85 (d 1H, J = 8 Hz), 8.12 (d 1H, J = 8 Hz), 6.76 (s 1H); m/z = 153.
3-Me-6-NO2 phenol: δ10.54 (s 1H, OH), 2.45 (s 3H, Me), 6.98 (s 1H), 7.98 (6, 1H, dd, J = 8 Hz), 6.92 (d 1H, J = 8 Hz); m/z = 153.
4-NO2-2-Cl phenol: δ10.66 (s 1H, OH), 7.15 (d 1H, J = 8.5 Hz), 8.12 (d 1H, J = 8.5 Hz), 8.34 (s 1H); m/z = 174.
6-NO2-2-Cl phenol: δ10.64 (s 1H, OH), 7.45 (dd 1H, J = 8 Hz, J = 7.5 Hz), 7.85 (d, 1H, J = 7.5 Hz), 8.16 (d 1H, J = 8 Hz); m/z = 174.
2-NO2-4-Cl phenol: δ10.54 (s 1H, OH), 7.12 (d 1H, J = 8 Hz), 7.82 (d 1H, J = 8 Hz), 8.36 (s 1H); m/z = 174.
2-NO2-4-Br phenol: δ10.45 (s 1H, OH), 7.10 (d 1H, J = 8 Hz), 7.72 (d 1H, J = 8 Hz), 8.25 (s 1H); m/z = 218.
2-NO2-benzene-1,4-diol: δ10.24–10.30 (s 2H, OH), 7.14 (d 1H, J = 8.5 Hz), 6.92 (d 1H, J = 8.5 Hz), 7.48 (s 1H); m/z = 155.
2-NO2-1-naphthol: δ12.24 (s 1H, OH), 7.66 (m, 1H, J = 8 Hz, J = 7.25 Hz), 7.76 (m, 1H, J = 8.5 Hz, J = 7.25 Hz), 8.58 (d 1H, J = 8.5 Hz), 8.05 (d 1H, J = 8.5 Hz, J = 4 Hz), 8.153 (d 1H, J = 8.5 Hz), 7.44 (d 1H, J = 9 Hz); m/z = 189.
1-NO2-2-naphthol: δ12.18 (s 1H, OH), 7.58 (m 1H, J = 7.5 Hz, J = 7.25 Hz), 7.80 (m 1H, J = 8.25 Hz, J = 7.25 Hz), 7.20 (d 1H, J = 9 Hz), 8.10 (m 1H, J = 7.75 Hz, J = 5 Hz), 7.68 (8, 1H, J = 8.25 Hz, J = 5 Hz), 8.65 (d, 1H, J = 9 Hz); m/z = 189.
2,4-dinitro-1-napthol: δ12.38 (s 1H, OH), 7.62 (m 1H, J = 8.5 Hz, J = 7.5 Hz), 7.92 (m 1H, J = 8 Hz, J = 7.5 Hz), 8.64 (s 1H), 8.35 (d 1H, J = 8.5 Hz), 8.22 (d 1H, J = 8 Hz); m/z = 234.
Conclusion
In conclusion, we have demonstrated that ultrasonic and microwave-assisted nitration reactions underwent smoothly with metal nitrates in the presence of PEGs. These methods have several advantages over existing methods such as regio-selectivity, high yields, simple procedure, and short reaction times. The observed reaction times are in the range of 40–45 min under sonication, 3–5 min under microwave-assisted conditions.
Supplemental data and research materials
Supplemental data for this article can be accessed at the Taylor & Francis website, doi: 10.1080/17518253.2015.1105309
Supplemental_Material.docx
Download MS Word (36.3 KB)Acknowledgements
The authors are indebted to Professor P.K. Saiprakash (Former Dean, Faculty of Science, OU), Professor T. Navaneeth Rao (Former Vice-Chancellor, OU), Principal, Nizam College and Head, Department of Chemistry, OU for constant encouragement and facilities.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
The authors would sincerely thank UGC and CSIR (New Delhi) for financial support in the form of JRF, accorded to B. Suresh, V. Sudhakar Chary, P. Srinivas, and G. Krishnaiah, which has become a driving force to carry out this work.
References
- Olah, G.A.; Malhotra, R.; Narang, S.C. Organic Nitro Chemistry Series – Nitration Methods and Mechanisms; VCH: New York, 1989.
- Hoggett, J.G.; Monodie, R.B.; Penton, J.R.; Schofield, K. Nitration and Aromatic Reactivity; Cambridge University Press: London, 1971.
- Ono, N. The Nitro Group in Organic Synthesis; Wiley-VCH: New York, 2001.
- Paul Selvam, J.J.; Suresh, V.; Rajesh, K.; Ravinder Reddy, S.; Venkateswarlu, Y. Tetrahedron Lett. 2006, 47, 2507–2509.
- Kogelbauer, A.; Vassena, D.; Prins, R.; Armor, J.N. Catal. Today. 2000, 55, 151–160.
- Riego, J.M.; Sedin, Z.; Zaldivar, J.M.; Marziano, N.C.; Tortato, C. Tetrahedron Lett. 1996, 37, 513–516.
- Peng, X.; Suzuki, H.; Lu, C. Tetrahedron Lett. 2001, 42, 4357–4359.
- Radoslaw, R.B.; Andrew, J.S. Tetrahedron Lett. 2001, 42, 6767–6769.
- Rodrigues, J.A.R.; Oliveira Filho, A.P.; Moran, P.J.S.; Custodio, R. Tetrahedron. 1999, 55, 6733–6738.
- Firouzabadi, H.; Iranpoor, N.; Zolfigol, M.A. Iran. J. Chem. Chem. Eng. 1997, 16, 48–58.
- (a) Anastas, P.T.; Warner, J. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, 1998; (b) Anastas, P.T.; Williamson, T.C. Green Chemistry: Frontiers in Benign Chemical Synthesis and Processes; Oxford University Press: New York, 1998; (c) Lancaster, M. Green Chemistry: An Introductory Text; RSC: London, 2002; (d) Anastas, P.T.; Kirchhoff, M.M. Acc. Chem. Res. 2002, 35, 686–694; (e) Lancaster, M. Green Chemistry: An Introductory Text; RSC: Cambridge, 2002; (f) Yamamoto, A. Pure. Appl. Chem. 2002, 74, 1–5; (g) Eissen, M.; Metzger, J.O. Chem. Eur. J. 2002, 8, 3580–3585; (h) Andrade, Carlos Kleber Z.; Alves, Luana M. Curr. Org. Chem. 2005, 9, 195–218; (i) Li, C.J. Chem. Rev. 2005, 105, 3095–3166.
- Kobayashi, Shū. Water in Organic Synthesis, Workbench Edition (Science of Synthesis); 1st ed., Georg Thieme Verlag: Stuttgart, 2012.
- (a) Rideout, D.C.; Breslow, R. J. Am. Chem. Soc. 1980, 102, 7816–7817; (b) Breslow, R.; Maitra, U.; Rideout, D.C. Tetrahedron Lett. 1983, 24, 1901–1904; (c) Breslow, R.; Maitra, U. Tetrahedron Lett. 1984, 25, 1239–1240.
- (a) Grieco, P.A.; Garner, P.; He, Z. J. Org. Chem. 1983, 24, 1897–1900; (b) Grieco, P.A.; Yoshida, K.; Garner, P. J. Org. Chem. 1983, 48, 3137–3139.
- Lindström, U.M. Chem. Rev. 2002, 102, 2751–2772.
- Corma, A.; Garcia, H. Chem. Rev. 2003, 103, 4307–4366.
- Francisco Alonso, Irina P. Beletskaya; Yusa, Miguel. Tetrahedron. 2005, 61, 11771–11835.
- (a) Dittmer, D.C. Chem. Ind. 1997, 19, 779–784; (b) Kumar, A.; Sharma, S. Green Chem. 2011, 13, 2017–2020.
- (a) Mason, T.J. Chemistry with Ultrasound; Elsevier Science Publishers Ltd.: London, 1990; (b) Kottrorearou, A.; Hoffman, M.R. J. Phys. Chem. 1991, 95, 3630–3638; (c) Suslick, K.S.; Flannigan, D.J. Annu. Rev. Phys. Chem. 2008, 59, 659–683; (d) Suslick, K.S. Ultrasound, Its Chemical, Physical and Biological Effects; VCH: New York, 1988.
- (a) Margulis, M.A. Advances in Sonochemistry. In Greenwich Connection, Vol. 1: Mason, T.J., Ed.; JA: London, 1990; p. 49; (b) Mason, T.J. Chem. Soc. Rev. 1997, 26, 443–451.
- (a) Suslick, K.S. Science, 1990, 4, 1439–1445; (b) Cravotto, G.; Cintas, P. Chem. Soc. Rev. 2006, 35, 180–196; (c) Fillion, H.; Luche, J.L. Selected Experiments. In Synthetic Organic Sonochemistry, Luche, J.L., Ed., Plenum: New York, 1998; pp. 91–106.
- (a) Goharshadi, E.K.; Ding, Y.; Jorabachi, N.M.; Nancarrow, P. Ultrason. Sonochem. 2009, 16, 120–123; (b) Mahdavinia, G.H.; Rostamizadeh, S.; Amani, A.M.; Emdadi, Z. Ultrason. Sonochem. 2009, 16, 7–10; (c) Puri, S.; Kaur, B.; Parmar, A.; Kumar, H. Ultrason. Sonochem. 2009, 16, 705–707.
- Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Tetrahedron. 2001, 5, 9225–9283.
- (a) Varma, R.S. Green Chem. 1999, 1, 43–55; (b) Vivek, P.; Varma, R.S. Chem. Res. 2008, 41, 629–639.
- Oliver Kappe, C. Angew. Chem. Int. Ed. 2004, 43, 6250–6284.
- Strauss, C.R.; Trainor, R.W. Aust. J. Chem. 1995, 48, 1665–1692.
- Abraham, M.A.; Moens, L. Clean Solvents, Alternative Media for Chemical Reactions and Processing, ACS Symposium Series 819; American Chemical Society: Washington, DC, 2002.
- Chen, J.; Spear, S.K.; Huddleston, J.G.; Rogers, R.D. Green Chem. 2005, 7, 64–82.
- (a) Ferravoschi, P.; Fiecchi, A.; Grisenti, P.; Santaniello, E.; Trave, S. Synth. Commun. 1987, 17, 1569–1575; (b) Leininger, N.F.; Clontz, R.; Gainer, J.L.; Kirwan, D.J. Chem. Eng. Commun. 2003, 190, 431–444; (c) Sukata, K. Bull. Chem. Soc. Jpn. 1984, 57, 613–614; (d) Badone, D.; Jommi, G.; Pagligrin, R.; Tavecchia, P. Synthesis 1987, 1987 (10), 920–921; (e) Chandrasekhar, S.; Narsihmulu, Ch.; Sultana, S.S.; Reddy, N.R. Chem. Commun. 2003, No. 14, 1716–1717; (f) Chandrasekhar, S.; Narsihmulu, Ch.; Chandrashekar, G.; Shyamsunder, T. Tetrahedron Lett. 2004, 45, 2421–2423; (g) Santaniello, E.; Manzocchi, A.; Sozzani, P. Tetrahedron Lett. 1979, 20, 4581–4582.
- (a) Wei, T.B.; Chen, J.C.; Wang, X.C.; Zhang, Y.M.; Wang, L.L. Synth. Commun. 1996, 26, 1447–1454; (b) Wang, L.L.; Lu, S.J.; Li, S.B.; Zhang, Y.M.; Wei, T.B. Synth. Commun. 1998, 28, 1005–1011.
- (a) Pearson, R.G. Chemical Hardness; John Wiley-VCH: Weinheim, 1997; (b) Pearson, R.G. J. Am. Chem. Soc. 1963, 85, 3533–3539; (c) Pearson, R.G. J. Chem. Educ. 1968, 45, 581–586.
- (a) Vogel, A.I. Text Book of Practical Organic Chemistry; 4th ed. Longman: London, 1986; (b) David R.L. CRC Handbook of Chemistry and Physics; 83rd ed., Chemical Rubber Company: Cleveland, Ohio, 2002–2003.