
ABSTRACT
We reported a green and simple Cu-catalyzed method for the efficient synthesis of 2-chloro-7-cyclopentyl-N,N-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide as the key intermediate in the synthetic approaches to pyrrolo[2,3-d]pyrimidine derivatives from 5-bromo-2,4-dichloropyrimidine through two routes in four steps and five steps, respectively. This method provided green and economical approaches toward numerous pyrrolo[2,3-d]pyrimidine derivatives.
GRAPHICAL ABSTRACT
Introduction
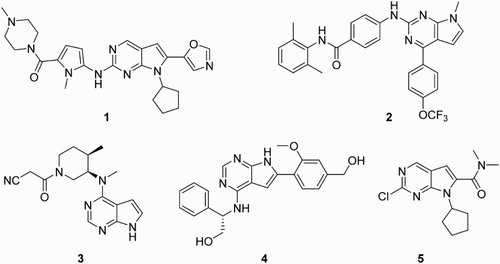
Pyrrolo[2,3-d]pyrimidine compounds, regarded as an important class of N-heterocycles, have played a significant role in medicinal chemistry ( Citation1–7) and material chemistry ( Citation8–10). Pyrrolo[2,3-d]pyrimidine skeleton is present in diverse naturally occurring products and synthesized bioactive compounds, most of which exhibit valuable bioactivities and fluorescence, as shown in . Some examples are as follows: aurora kinase inhibitor 1 that was developed by Brazidec et al. ( Citation11); hedgehog signaling pathway inhibitor 2 was achieved by Wei et al. ( Citation12), suggesting approximate threefold more effective than positive control GDC-0449; Tofacitinib 3 ( Citation13), which was developed by Pfizer, acted as a small-molecule pan-JAK inhibitor; EGFR-TK inhibitor 4 ( Citation14), which was discovered with higher potency in enzymatic assays than commercial drug Erlotinib.
Figure 1. Representative compounds.
2-Chloro-7-cyclopentyl-N,N-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide 5 was the key intermediate in the synthetic approaches to pyrrolo[2,3-d]pyrimidine derivatives (Citation15–18). Therefore, it has stimulated considerable efforts toward exploring efficient routes to the significant compound 5. Two methods have been previously reported by Besong ( Citation15) and Calienni ( Citation17), respectively. Besong used cyclopentylamine, 5-bromo-2,4-dichloropyrimidine and 3,3-diethoxy-propyne as starting materials to prepare compound 5 in six steps (). Besong's synthetic approaches to compound 5 included the selective N-alkylation of 5-bromo-2,4-dichloropyrimidine with cyclopentylamine to afford 5-bromo-2-chloro-N-cyclopentylpyrimidin-4-amine 6, followed by a Pd-catalyzed Sonogashira coupling with 3,3-diethoxy-propyne to yield the desired product 7 with moderate overall yield. The cyclization of the compound 7 with TBAF gave compound 8, which was converted into the corresponding aldehyde 9 by hydrolysis using hydrochloric acid. The oxidation of 9 with oxone gave the corresponding acid 10, which was transformed into the target amide 5 by the condensation reaction using HBTU, DIEA and dimethylamine. Compared with Besong’s method, Calienni replaced 3,3-diethoxy-propyne with propargyl alcohol. The target product 5 was prepared in four steps (). The first three steps of Calienni’s method were similar to that of Besong’s method. The fourth step was the preparation of compound 5 from compound 12 with dimethylamine, manganese (IV) oxide and sodium cyanide.
Scheme 1. Reagents and conditions: (a) cyclopentylamine, DIEA, EtOH, r.t., 89%; (b) 3,3-diethoxy-propyne, CuI, PdCl2(PPh3)2, Et3N, DMF, 100°C, 43%; (c) TBAF, THF, 65°C, 82%; (d) HCl concn, dioxane, r.t., 82%; (e) Oxone, DMF, r.t., 85%; and (f) HBTU, DIEA, dimethylamine, DMF, 79%.
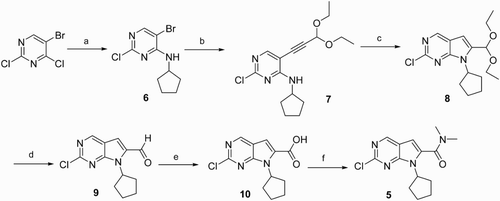
Scheme 2. Reagents and conditions: (a) cyclopentylamine, DIPEA, EtOAc, r.t., 88%; (b) propargyl alcohol, TBAF, PdCl2(PPh3)2, THF, 67°C, 47%; (c) TBAF, THF, 60°C, 82%; and (d) dimethylamine, NaCN, MnO2, DMF, 65%.
Results and discussion
According to Besong’s method and Calienni’s method, pyrrolo[2,3-d]pyrimidine derivatives were synthesized from 5-bromo-2,4-dichloropyrimidine and terminal alkynes through the Pd-catalyzed Sonogashira reaction and a followed tandem cyclization using tetrabutylammonium fluoride in two steps manner. The costly/toxic Pd catalyst was essential in the method. The pyrrolo[2,3-d]pyrimidine derivatives were prepared in moderate yield by using Pd. Recently, Cu-catalyzed coupling reactions devoted to the efficient synthesis of heterocycles have acquired increasing attention, and displayed lower cost/toxicity of Cu sources, mild conditions, and broad substrate scope ( Citation19–23). Ma et al. reported the first Cu/6-methylpicolinic acid catalyzed synthesis of pyrrolo[2,3-d]pyrimidine derivatives in moderate to excellent yields from substituted 5-bromopyrimidin-4-amines and alkynes ( Citation24). The method provides one-pot protocol toward affording a variety of pyrrolo[2,3-d]pyrimidines. Therefore, we conceived using the Cu/6-methylpicolinic acid system to develop a facile, practicable, and economical method for the synthesis of 2-chloro-7-cyclopentyl-N,N-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide 5.
In this contribution, we developed a convenient method for the preparation of compound 5. The synthetic route 1 (), which was based on Besong’s method, started with the reaction of 5-bromo-2,4-dichloropyrimidine and cyclopentylamine in N,N-diisopropylethylamine to yield compound 6, and then Cu/6-methylpicolinic acid catalyzed Sonogashira reaction/cyclization of compound 6 with 3,3-diethoxy-propyne to afford compound 8 in 85% yield. The hydrolysis of the compound 8 with hydrochloric acid generated aldehyde 9. Further oxidation of compound 9 with oxone gave the corresponding acid 10. The reaction of the compound 10 with SOCl2 provided the corresponding acid chloride, which was converted into the desired compound 5 by subsequent condensation condition using dimethylamine. Compared with Besong’s method, route 1 replaced the Pd catalysts with Cu catalysts. In this work, the 42% overall yield of route 1 in five steps was obtained, which was remarkably higher than the 17% overall yield of Besong’s method, because the Cu-catalyzed effect is better than Pd.
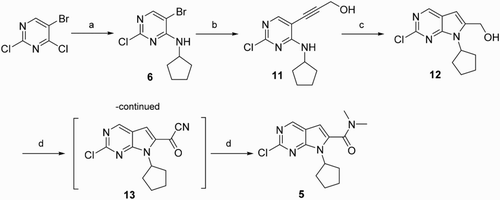
Scheme 3. Reagents and conditions: (a) cyclopentylamine, DIPEA, EtOAc, r.t., 88%; (b1) 3,3-diethoxy-propyne, CuCl, 6-methylpicolinic acid, NaI, K2CO3, DMSO; (b2) propargyl alcohol, CuCl, 6-methylpicolinic acid, NaI, K2CO3, DMSO; (c) HCl concn, THF, r.t., 89%; (d) oxone, DMF, r.t., 85%; (e) SOCl2, Dimethylamine, 76%; and (f) NaClO2, TEMPO, NaClO, CH3CN, sodium phosphate buffer (pH = 6.7), 35°C, 80%.
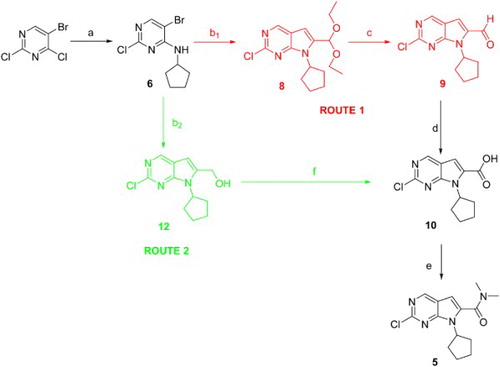
Because 3,3-diethoxy-propyne was replaced with propargyl alcohol and the oxidation of alcohol 12 to acid 10 was modified by reference to Calienni’s method and the synthetic route 1, the synthetic route 2 was developed. As depicted in Scheme 3, the N-arylation reaction of cyclopentylamine with 5-bromo-2,4-dichloropyrimidine in N,N-diisopropylethylamine yieldedcompound 6, and then Cu/6-methylpicolinic acid catalyzed Sonogashira reaction/cyclization of the compound 6 with propargyl alcohol gave the compound 12 in 60% yield, after the oxidation of the alcohol 12 using stoichiometric NaClO2, catalytic TEMPO, and catalytic NaClO to afford the corresponding acid 10 ( Citation25), the condensation condition of dimethylamine and the compound 10 was similar to that of route 1. The overall yield of route 2 from cyclopentylamine, 5-bromo-2,4-dichloropyrimidine and propargyl alcohol to compound 5 via four steps was 31%, which was lower than the yield of the route 1, because the debrominated compound of 6 as the major side product was obtained with 30% yield in Cu-catalyzed coupling reaction. Nevertheless, it was noteworthy that route 2 was one step shorter than route 1, because the alcohol was directly oxidized to the acid in one step. Compared with Calienni’s method, the advantages of route 2 included higher overall yield by using Cu catalyst and the one-pot, environmentally benign and efficient oxidation of primary alcohols to carboxylic acids using NaClO2/TEMPO/NaClO. As a comparison, Calienni’s method by using excessive amounts of manganese (IV) oxide as oxidant and sodium cyanide as reagent was not considered as an eco-friendly method.
Conclusion
In summary, we have developed a general, mild and environmentally benign method for the synthesis of 2-chloro-7-cyclopentyl-N,N-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide 5 through two routes with 42% and 31% overall yield, respectively. Such new routes have merits of facile reaction condition, inexpensive reagents, short steps, convenient operation and high yield. Due to the more efficient catalytic performance, Cu was used as catalyst instead of the costly/toxic Pd in this work. The methods provide green and practicable pathway toward other similar compounds having pyrrolo[2,3-d]pyrimidine skeleton.
Experimental
Unless otherwise stated, all reagents were purchased from commercial suppliers and used without further purification. DMSO was dried by activated 4 Å molecular sieve. CuCl was purified by concentrated hydrochloric acid. Column chromatography was performed with silica gel (200–300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. Thin-layer chromatography was carried out with Merck silica gel GF254 plates. All products were characterized by 1H NMR and 13C NMR and mass spectra (ESI). 1H NMR and 13C NMR spectra were recorded at room temperature on Bruker Avance IIIHD (400 MHz). Mass spectra were recorded on Agilent Q-TOF6530.
Route 1
Synthesis of 5-bromo-2-chloro-N-cyclopentylpyrimidin-4-amine (6).
To a solution of 5-bromo-2,4-dichloropyrimidine (2.28 g, 10 mmol, 1.0 eq.) in EtOAc (15 mL) was added N,N-diisopropylethylamine (2.59 g, 20 mmol, 2.0 eq.) and cyclopentylamine (1.28 g, 15 mmol, 1.5 eq.) with efficient stirring in 100 mL flask at r.t. The solution was stirred overnight. The solvent was evaporated and the crude residue was purified using silica gel chromatography (PE/EA = 10/1) to give 5-bromo-2-chloro-N-cyclopentylpyrimidin-4-amine 6 as a white solid (2.34 g, 85%). 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 5.46 (s, 1H), 4.42 (sextet, J = 7.0 Hz, 1H), 2.19–2.07 (m, 2H), 1.81–1.61 (m, 4H), 1.53–1.41 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 159.4, 159.0, 156.2, 103.0, 53.2, 33.1, 23.7. ESI-MS: m/z = 276.0 [M+H]+, m.p.: 95–96°C.
Synthesis of 2-chloro-7-cyclopentyl-6-(diethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (8).
A Schlenk tube was charged with 6 (2.2 g, 8 mmol), CuCl (80 mg, 0.8 mmol), 6-methylpicolinic acid (330 mg, 2.4 mmol), NaI (1.2 g, 8 mmol), and K2CO3 (3.3 g, 24 mmol). The tube was evacuated and refilled with argon three times before the solution of 3,3-diethoxyprop-1-yne (2.1 g, 16 mmol) in DMSO (16 mL) was added. The reaction mixture was stirred at 115°C for 24 h. After the reaction mixture was cooled to room temperature, saturated NH4Cl solution (100 mL) was added. The mixture was extracted with EtOAc (3 × 70 mL) and the organic layer was washed with water, brine and dried over Na2SO4. After concentration in vacuo, the residue was purified by column chromatography on silica gel (PE/EA = 10/1) to provide 2-chloro-7-cyclopentyl-6-(diethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine 8 as a white solid (2.2 g, 85%). 1H NMR (600 MHz, CDCl3) δ 8.73 (s, 1H), 6.62 (s, 1H), 5.65 (s, 1H), 4.98 (quintet, J = 9.0 Hz, 1H), 3.67–3.53 (m, 4H), 2.49–2.38 (m, 2H), 2.18–2.08 (m, 2H), 2.06–1.99 (m, 2H),1.75–1.64 (m, 2H), 1.25 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 152.7, 152.7, 150.8, 139.7, 117.6, 99.4, 96.7, 61.8, 57.4, 30.9, 25.2, 15.2. ESI-MS: m/z = 324.1 [M+H]+, m.p.: 65–66°C.
Synthesis of 2-chloro-7-cyclopentyl-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde (9).
To a solution of 8 (1.6 g, 5 mmol) in THF was added concentrated hydrochloric acid (1.5 mL). After the reaction was completed in 1 h, water was added and then the mixture was extracted with EtOAc. The organic layer was washed with water, brine and dried over Na2SO4. After concentration in vacuo, the residue was purified by column chromatography on silica gel (PE/EA = 3/1) to provide 2-chloro-7-cyclopentyl-7H-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde 9 (1.1 g, 89%). 1H NMR (400 MHz, CDCl3) δ 9.93 (s, 1H), 9.00 (s, 1H), 7.33 (s, 1H), 5.82–5.70 (m, 1H), 2.35–2.20 (m, 2H), 2.20–2.03 (m, 4H), 1.80–1.65 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 182.4, 156.8, 155.0, 154.1, 137.6, 116.5, 116.0, 56.9, 31.3, 25.0. ESI-MS: m/z = 250.1 [M+H]+, m.p.: 162–164°C.
Synthesis of 2-chloro-7-cyclopentyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (10).
To a solution of 9 (1.25 g, 5 mmol) in DMF was added oxone (3.6 g, 6 mmol) and stirred for 6 h. After the reaction was completed, water was added and white solid was precipitated and filtered to give 2-chloro-7-cyclopentyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid 10 (1.13 g, 85%). 1H NMR (400 MHz, DMSO) δ 13.71 (br. s, 1H), 9.07 (s, 1H), 7.37 (s, 1H), 5.85– 5.65 (m, 1H), 2.35–2.20 (m, 2H), 2.07–1.93 (m, 4H), 1.70–1.57 (m, 2H).13C NMR (100 MHz, DMSO) δ 162.0, 154.7, 153.8, 152.4, 131.6, 116.3, 108.2, 56.2, 30.4, 24.7. ESI-MS: m/z = 266.1 [M+H]+, m.p.: 223–225°C.
Synthesis of 2-chloro-7-cyclopentyl-N,N-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide (5).
To a slurry of 10 (1 g, 4 mmol) in CH2Cl2 (15 mL) was added SOCl2 (2 mL) and DMF (0.2 mL) with efficient stirring in 50 mL flask at r.t. The solution was stirred for 2 h. Solvent was evaporated and the crude residue was dissolved in CH2Cl2 (15 mL). Then the solution was added dropwise into ice-cold solution of dimethylamine in THF (5 mL, 2 N) maintained 0°C with efficient stirring. Then the solution was stirred for 2 h at r.t. Solvent was evaporated and the crude residue was purified using silica gel chromatography (PE/EA/Et3N = 1/1/3‰) to give 2-chloro-7-cyclopentyl-N,N-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide 5 as a white solid (888 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 8.77 (s, 1H), 6.51 (s, 1H), 4.93–4.77 (m, 1H), 3.15 (s, 3H), 3.07 (s, 3H), 2.40–2.25 (m, 2H), 2.13–1.95 (m, 4H), 1.75–1.55 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 163.4, 153.7, 152.2, 151.9, 135.6, 117.2, 99.8, 58.0, 39.3, 35.2, 31.1, 24.9. ESI-MS: m/z = 293.1 [M+H]+, m.p.: 105–106°C.
Route 2
Synthesis of (2-chloro-7-cyclopentyl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)methanol (12).
A Schlenk tube was charged with 6 (2.2 g, 8 mmol), CuCl (80 mg, 0.8 mmol), 6-methylpicolinic acid (330 mg, 2.4 mmol), NaI (2.4 g, 16 mmol), and K2CO3 (3.3 g, 24 mmol). The tube was evacuated and refilled with argon three times before the solution of propargyl alcohol (1.8 g, 32 mmol) in DMSO (16 mL) was added. The reaction mixture was stirred at 100°C for 48 h. After the reaction mixture was cooled to room temperature, saturated NH4Cl solution (100 mL) was added. The mixture was extracted with EtOAc (3 × 70 mL) and the organic layer was washed with water, brine and dried over Na2SO4. After concentration in vacuo, the residue was purified by column chromatography on silica gel (PE/EA = 2/1) to provide (2-chloro-7-cyclopentyl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)methanol 12 as a white solid (1.2 g, 60%).1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 6.43 (s, 1H), 4.97–4.88 (m, 1H), 4.83 (s, 2H), 2.50–2.30 (m, 2H), 2.20–2.00 (m, 4H), 1.80–1.63 (m, 2H). 13C NMR (100 MHz, DMSO) δ 151.9, 151.0, 150.5, 144.2, 117.7, 98.1, 56.0, 55.8, 30.3, 24.5. ESI-MS: m/z = 252.1 [M+H]+, m.p.: 176–178°C.
Synthesis of 2-chloro-7-cyclopentyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (10).
A mixture of 12 (2 g, 8 mmol), TEMPO (200 mg, 1.28 mmol), CH3CN (120 mL), and sodium phosphate buffer (80 mL, 0.67 M, pH = 6.7) was heated to 35°C. Then sodium chlorite (4 g in 16 mL water) and dilute bleach (0.2 mL diluted into 8 mL) were added simultaneously over 45 min. The mixture was stirred at 35°C until the reaction is completed (10 h), and then cooled to room temperature. Water was added, and the pH was adjusted to 8.0 with 2.0 N NaOH. The reaction was quenched by pouring into ice-cold Na2SO3 solution maintained below 20°C. The pH of the aqueous layer should be 8.5–9.0. After stirring for 0.5 h at room temperature, the aqueous layer was acidified with 2.0 N hydrochloric acid. The mixture was extracted with EtOAc and the organic layer was washed with water, brine and dried over Na2SO4. After concentration in vacuo, the residue was purified by column chromatography on silica gel (CH2Cl2/MeOH/HCOOH = 30/1/3‰) to provide 2-chloro-7-cyclopentyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid 10 as a white solid (1.7 g, 80%).1H NMR (400 MHz, DMSO) δ 13.71 (br. s, 1H), 9.07 (s, 1H), 7.37 (s, 1H), 5.85–5.65 (m, 1H), 2.35–2.20 (m, 2H), 2.07–1.93 (m, 4H), 1.70–1.57 (m, 2H). 13C NMR (100 MHz, DMSO) δ 162.0, 154.7, 153.8, 152.4, 131.6, 116.3, 108.2, 56.2, 30.4, 24.7. ESI-MS: m/z = 266.1 [M+H]+, m.p.: 223–225°C.
Supplement_material.doc
Download MS Word (691 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Dr. Leilei Wang, received his bachelor’s degree in Applied Chemistry from Qufu Normal University, Qufu, China, in 2007 and doctor’s degree in Organic Chemistry from Sun Yat-Sen University, Guangzhou, China, in 2013, respectively. Recently, he is working as “assistant professor” in Ecology institute, Shandong Academy of Sciences, Jinan, China.
Liwen Zheng, received his bachelor’s degree in Chemistry from Yantai Normal University, Yantai, China, in 2005 and master's degree in Organic Chemistry from Zhengzhou University, Zhengzhou, China, in 2008, respectively. Recently, he is working as “assistant professor” in Ecology institute, Shandong Academy of Sciences, Jinan, China.
Xue Kong, received her bachelor’s degree in Applied Chemistry from China University of Petroleum, Dongying, China, in 2001 and master’s degree in Applied Chemistry from China University of Petroleum, Dongying, China, in 2004, respectively. Recently, she is working as “associate professor” in Ecology institute, Shandong Academy of Sciences, Jinan, China.
Wen Zhang, received her bachelor’s in Environmental Science from Wuhan University of Science & Technology, Wuhan, China, in 2006 and doctor’s degree in Environmental Science from Nankai University, Tianjin, China, in 2011, respectively. Recently, she is working as “assistant professor” in Ecology institute, Shandong Academy of Sciences, Jinan, China.
Guanhong Chen, received her bachelor’s in Pharmacy from The Second Military Medical University, Shanghai, China, in 1996 and master’s degree in Environmental Chemistry from Nanjing Tech University, Nanjing, China, in 2001, respectively. Recently, she is working as “associate professor” in Ecology institute, Shandong Academy of Sciences, Jinan, China.
Jianing Wang, received his bachelor’s degree in Chemical Engineering from Tianjin University, Tianjin, China, in 1989 and doctor’s degree in Chemical Technology from Tianjin University, Tianjin, China, in 2002, respectively. Recently, he is working as “professor” in Ecology institute, Shandong Academy of Sciences, Jinan, China.
Additional information
Funding
Related Research Data
References
- Murphy, C. G.; Dickler, M. N. Oncologist. 2015, 20, 483–490. doi: 10.1634/theoncologist.2014-0443
- Mohamed, M. S.; El-Hameed, R. A.; Sayed, A. I.; Soror, S. H. Arch. Pharm. 2015, 348, 194–205. doi: 10.1002/ardp.201400387
- Jiao, X. Y.; Kopecky, D. J.; Liu, J. S.; Liu, J. Q.; Jaen, J. C.; Cardozo, M. G.; Sharma, R.; Walker, N.; Wesche, H.; Li, S.; Farrelly, E.; Xiao, S.-H.; Wang, Z.; Kayser, F. Bioorg. Med. Chem. Lett. 2012, 22, 6212–6217. doi: 10.1016/j.bmcl.2012.08.020
- Naus, P.; Caletkova, O.; Konecny, P.; Dzubak, P.; Bogdanova, K.; Kolář, M.; Vrbková, J.; Slavětínská, L.; Tloušt’ová, E.; Perlíková, P.; Hajdúch, M.; Hocek, M. J. Med. Chem. 2014, 57, 1097–1110. doi: 10.1021/jm4018948
- Kumar, V. P.; Frey, K. M.; Wang, Y.; Jain, H. K.; Gangjee, A.; Anderson, K. S. Bioorg. Med. Chem. Lett. 2013, 23, 5426–5428. doi: 10.1016/j.bmcl.2013.07.037
- Devine, S. M.; Lim, S. S.; Chandrashekaran, I. R.; MacRaild, C. A.; Drew, D. R.; Debono, C. O.; Lam, R.; Anders, R. F.; Beeson, J. G.; Scanlon, M. J.; Scammells, P. J.; Norton, R. S. Med. Chem. Commun. 2014, 5, 1500–1506. doi: 10.1039/C4MD00090K
- Zhao, X.; Huang, W.; Wang, Y.; Xin, M.; Jin, Q.; Cai, J.; Tang, F.; Zhao, Y.; Xiang, H. Bioorg. Med. Chem. 2015, 23, 891–901. doi: 10.1016/j.bmc.2014.10.043
- Tumkevicius, S.; Dodonova, J.; Kazlauskas, K.; Masevicius, V.; Skardziute, L.; Jursenas, S. Tetrahedron Lett. 2010, 51, 3902–3906. doi: 10.1016/j.tetlet.2010.05.093
- Skardziute, L.; Kazlauskas, K.; Dodonova, J.; Bucevicius, J.; Tumkevicius, S.; Jursenas, S. Tetrahedron. 2013, 69, 9566–9572. doi: 10.1016/j.tet.2013.09.044
- Bucevicius, J.; Skardziute, L.; Dodonova, J.; Kazlauskas, K.; Bagdziunas, G.; Jursenas, S.; Tumkevicius, S. RSC Adv. 2015, 5, 38610–38622. doi: 10.1039/C5RA05482F
- Brazidec, J.-Y. L.; Pasis, A.; Tam, B.; Boykin, C.; Wang, D.; Marcotte, D. J.; Claassen, G.; Chong, J.-H.; Chao, J.; Fan, J.; Nguyen, K.; Silvian, L.; Ling, L.; Zhang, L.; Choi, M.; Teng, M.; Pathan, N.; Zhao, S.; Li, T.; Taveras, A. Bioorg. Med. Chem. Lett. 2012, 22, 4033–4037. doi: 10.1016/j.bmcl.2012.04.085
- Zhang, L.; Xin, M.; Shen, H.; Wen, J.; Tang, F.; Tu, C.; Zhao, X.; Wei, P. Bioorg. Med. Chem. Lett. 2014, 24, 3486–3492. doi: 10.1016/j.bmcl.2014.05.066
- Clark, J. D.; Flanagan, M. E.; Telliez, J. B. J. Med. Chem. 2014, 57, 5023–5038. doi: 10.1021/jm401490p
- Sundby, E.; Han, J.; Kaspersen, S. J.; Hoff, B. H. Eur. J. Pharm. Sci. 2015, 80, 56–65. doi: 10.1016/j.ejps.2015.08.003
- Besong, G.; Brain, C. T.; Brooks, C. A.; Congreve, M. S. CN 102186856, 2010.
- Brain, C. T.; Cho, Y. S.; Giraldes, J. W.; Lagu, B. WO 2011/101409, 2011.
- Calienni, J. V.; Chen, G.-P.; Gong, B.; Kapa, P. K.; Saxena, V. US 2012/0115878, 2012.
- Borland, M.; Brain, C. T.; Doshi, S.; Kim, S. WO 2011/130232, 2011.
- Evano, G.; Blanchard, N. Copper-Mediated Cross-coupling Reaction. John Wiley & Sons, Inc.: Hoboken, NJ, 2014.
- Monnier, F.; Taillefer, M. Angew. Chem. Int. Ed. 2009, 48, 6954–6971. doi: 10.1002/anie.200804497
- Liu, Y.; Wan, J. P. Org. Biomol. Chem. 2011, 9, 6873–6894. doi: 10.1039/c1ob05769c
- Thapa, S.; Shrestha, B.; Gurung, S. K.; Giri, R. Org. Biomol. Chem. 2015, 13, 4816–4827. doi: 10.1039/C5OB00200A
- Guo, X.; Gu, D.-W.; Wu, Z.; Zhang, W. Chem. Rev. 2015, 115, 1622–1651. doi: 10.1021/cr500410y
- Jiang, X.; Sun, D.; Jiang, Y.; Ma, D. Tetrahedron Lett. 2015, 56, 3259–3261. doi: 10.1016/j.tetlet.2014.12.098
- Zhao, M.; Li, J.; Mano, E.; Song, Z.; Tschaen, D. M.; Grabowski, E. J. J.; Reider, P. J. J. Org. Chem. 1999, 64, 2564–2566. doi: 10.1021/jo982143y