
ABSTRACT
Proteolytic processing regulates key processes in health and disease. The cellular prion protein (PrPC) is subject to at least 3 cleavage events, α-cleavage, β-cleavage and shedding. In contrast to α- and β-cleavage where there is an ongoing controversy on the identity of relevant proteases, the metalloprotease ADAM10 represents the only relevant PrP sheddase. Here we focus on the roles that ADAM10-mediated shedding of PrPC and its pathogenic isoform (PrPSc) might play in regulating their physiological and pathogenic functions, respectively. As revealed by our recent study using conditional ADAM10 knockout mice (Altmeppen et al., Citation2015), shedding of PrP seems to be involved in key processes of prion diseases. These aspects and several open questions arising from them are discussed. Increased knowledge on this topic can shed new light on prion diseases and other neurodegenerative conditions as well.
Introduction
The cellular prion protein (PrPC) is a membrane-anchored glycoprotein that is highly expressed in neurons of the central and peripheral nervous system. The N-terminal part of PrPC is highly flexible and contains an octameric repeat region, a neurotoxic domain and a hydrophobic core. The C-terminal part is structured and comprises α-helices, β-strands, loop domains, 2 N-linked glycosylation sites and a glycosylphosphatidylinositol (GPI)-anchor for attachment to the outer leaflet of the plasma membrane.Citation1,2
Conformational conversion of PrPC into a misfolded isoform (PrPSc) is critically involved in initiation and progression of fatal neurodegenerative prion diseases such as Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cattle and chronic wasting disease (CWD) in deer.Citation3,4 PrPSc is regarded as an essential component of the transmissible entity of prion diseases, namely the prion, which appears to be devoid of nucleic acids.Citation5–7
Long before it has been realized that proteolytic cleavage of PrPC occurs constitutively under physiological and pathological conditions, PrP has been subjected to proteolytic processing for diagnostic and research purposes. In laboratories, artificial proteolysis of PrP is achieved by proteinase K to assess presence of PrPSc and to gain insight into its structural features.Citation8 In addition, release of full-length PrPC is accomplished by GPI-anchor cleavage upon treatment with phosphatidylinositol-specific phospholipase C (PI-PLC).Citation9,10
The relevance of naturally occurring cleavage events has only recently been realized and it seems that PrP cleavage regulates PrPC levels and functions. Moreover, generated PrP fragments are bioactive.Citation11–17 Here, we will focus on the release of nearly full-length PrPC by the metalloprotease ADAM10 while leaving other cleavage events, such as α-cleavage and β-cleavage, aside as these have recently been reviewed in detail.Citation18–20
Shedding of PrPC
Soluble forms of nearly full-length PrPC have been described in human CSFCitation21 and blood,Citation22,23 yet insight into the molecular mechanisms on the generation of these PrPC species were only gained recently. The release of extracellular domains of membrane-bound proteins by proteolytic cleavage is termed “shedding”. Shedding of PrPC was initially described by Borchelt et al. in primary cultures of neonatal Syrian hamster brain.Citation24 Additional experiments in primary lymphoid and neuronal cells and in cultured cell lines showed that this mechanism is conserved across cell types and species.Citation25,26 By using inhibitors of proteases, the first hints that the sheddase belongs to the family of metalloproteases have been followedCitation24,27 and finally ADAM10 was identified as the PrPC sheddase.Citation28 Digestion experiments in a cell-free system using recombinant proteins recently confirmed these findings.Citation29 Additionally, it has been shown that ADAM9 regulates the activity of ADAM10 and thus indirectly influences PrPC shedding.Citation30–32 The cleavage site could be mapped between Gly(228) and Arg(229) (in murine PrPC) thus locating merely 3 residues distant to the GPI-anchor attachment.Citation28,33 In contrast to the conclusive data concerning shedding of PrPC gained from cell culture studies, in vivo data regarding this important processing step are not as detailed. Transgenic mice moderately overexpressing ADAM10 have reduced PrPC levels and present with decreased PrPSc amounts and prolonged incubation times upon challenge with prions.Citation34
The availability of conditional ADAM10 knockout mouse models circumvented the problem of early embryonic lethality occurring in complete ADAM10 knockout miceCitation35 and enabled us to study the contribution of this protease to the shedding of PrPC in vivo.Citation35,36 Using 2 different conditional ADAM10 knockout mouse lines with deletion of ADAM10 in either neuronal precursorsCitation36 or forebrain neuronsCitation37, we were able to confirm ADAM10 as the relevant PrPC sheddase in vivo.Citation38,39
These mice gave further valuable insights into the effects that lack of PrPC shedding has in membrane homeostasis of PrPC and in the context of prion disease. Firstly, lack of ADAM10-mediated shedding leads to disturbed posttranslational processing and membrane homeostasis of PrPC at the neuronal plasma membrane, where—as a consequence— levels of PrPC increase. Secondly, due to impaired shedding, PrPC also accumulates in the early secretory pathway while its mRNA levels remain unaffected.
Shedding of PrPC in Prion Disease
Resistance of cells that express only anchorless PrP toward chronic prion infection has been demonstrated.Citation40 In line with this, cell culture based experiments gave evidence that forced artificial release of full-length PrPC by exposure to PtdIns-PLC not only prevents prion infection of susceptible cell lines but also cures chronically prion-infected cell lines from producing PrPSc.Citation41,42 In vivo data on the role of shedding of PrPC or PrPSc lagged behind when compared to data obtained from cell culture experiments. The first hints that soluble versions of PrPC have profound effects on the pathophysiology of prion disease came from transgenic mice expressing soluble PrPC dimers. In these mice, soluble PrPC dimers were able to antagonize PrPSc propagation and did not form PrPSc themselves.Citation43 Since in this model PrPC was fused to the relatively large Fcγ tail of human IgG1 (PrPC-Fc) and was dimerized, the data cannot be directly transferred to bona fide shed PrPC. Other transgenic mice expressing PrPC with a stop codon inserted C-terminal to the putative ADAM10 cleavage site more closely mimicked transgenic expression of shed PrPC.Citation44,45 In contrast to PrPC-Fc, anchorless PrPC can be converted to PrPSc and this results in an altered type of prion disease upon challenge with prions.Citation44,45 Later it turned out that higher expression levels of anchorless PrPC can even give rise to spontaneous generation of prions.Citation46 However, none of these studies investigated the role of physiological shedding of PrP in prion disease.
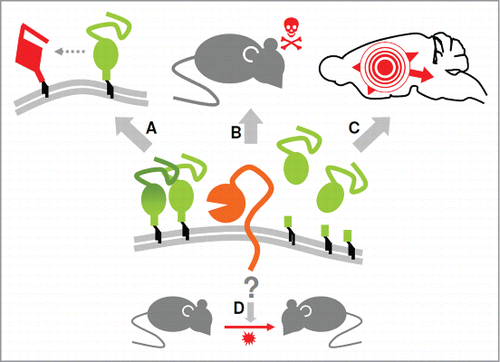
Our conditional forebrain-specific ADAM10 knockout (ADAM10 cKO) mice enabled us to perform prion inoculation experiments in mice lacking the ability to generate neuronally shed PrPC.Citation39 Briefly, besides elevated PrPC membrane levels, depletion of ADAM10 resulted in increased PrPSc formation and shortened incubation time to prion disease. While lack of ADAM10 did not influence prion infectivity our data suggested a role of ADAM10-mediated shedding in the spread of prion-associated pathology within the brain. Thus, findings obtained with this mouse model touch several aspects that are currently discussed in the prion field () and provide insight into several open questions discussed in more detail below.
Figure 1. ADAM10 influences several aspects of prion diseases. As revealed by our recent study using ADAM10 cKO mice (as a model for depleted neuronal shedding of PrP) and Tga20 mice (as a model for efficient PrP shedding) the sheddase ADAM10 controls membrane levels and production of anchorless PrP (center) and, by doing so, seems to influence PrPSc formation (A) as well as neurotoxicity and incubation times (B). Moreover, spread of prion-associated pathology throughout the brain appeared to be affected (C). In contrast, our study did not indicate involvement of the protease in the production of infectious prions and thus on transmissibility (D). Details are discussed in the main text.
The Different Facets of ADAM10-Mediated Shedding in Prion Disease
PrPC Membrane Levels and the Execution of Neurotoxicity in Prion Diseases
Expression of PrPC is an absolute requirement for the establishment of prion diseases.Citation47–49 The importance of GPI-anchored PrPC for both PrPSc conversion and neurotoxicity, 2 mechanistically different yet related aspects in prion diseases,Citation50,51 has been revealed by several studies (for a review see refs. Citation52–54).
However, it is not exactly clear how membrane-bound PrPC mediates neurotoxicity. PrPC acts as a receptor for various oligomeric β-sheet-rich protein species associated with different neurodegenerative diseases such as Alzheimer's (AD) and Parkinson's disease (PD).Citation15,55–57 Binding of these toxic conformers—in the case of prion diseases presumably specific assemblies of PrPSc—to PrPC at the neuronal surface may initiate signaling pathways ultimately leading to loss of synapses and neurons.Citation15,55,58–60 Another mechanism of neurotoxicity is thought to arise from membrane poresCitation61,62 formed either directly by PrPSc aggregates or by the N-terminus of PrPC upon structural changes.Citation63,64 Such pores would then disturb the neuronal ion homeostasis and calcium influx leading to activation of calpain and possibly uncontrolled downstream proteolysis. While we indeed observed a correlation between increased PrPSc amounts and upregulation of calpain indicative for increased pore formation in our ADAM10 cKO mice, we could not find evidence for activation of toxic signaling cascades supporting the receptor model.Citation39 However, this might have been overlooked in our study. To elucidate this point, a more detailed investigation might be necessary (e.g. analysis of synapotosome preparations). In fact, it is conceivable that more mechanisms of neurotoxicity exist and that they all act in concert in prion diseases.
Neurotoxicity seems to be the determinant of incubation times in prion disease and is likely dependent on the amount of PrPC at the plasma membrane. The connection between amounts of membrane-bound PrPC and survival times is strengthened by our study since levels of surface PrPC expression (Tga20 > ADAM10 cKO > wild type) inversely correlated with incubation times (wild type > ADAM10 cKO > Tga20).Citation39 Of note, lack of ADAM10-mediated shedding reduced the incubation time by approximately 30%Citation39, whereas overexpression of the protease leads to prolonged survival.Citation34 Additionally, in mice expressing anchorless PrPC, coexpression of membrane-bound PrPC results in accelerated clinical prion disease.Citation44,46
Taken together, these findings clearly support the view that maintenance of plasma membrane levels of PrPC is a critical factor for susceptibility toward prion infection and suggest ADAM10 as a key modulator.
Inhibition of PrPSc Formation by Shed PrP?
Conversion of PrPC to PrPSc is likely to occur at the cell surface, yet a considerable amount of newly formed PrPSc then rapidly traffics to intracellular compartments.Citation65,66 Our ADAM10 cKO mice showed significantly higher levels of PrPSc when compared to controlsCitation39 arguing that, as observed in cell culture based experiments,Citation65,66 also in vivo conversion of PrPC to PrPSc mainly occurs at the plasma membrane. However, a connection between surface PrPC levels and efficiency of PrPSc formation cannot be generalized since Tga20 mice, despite increased PrPC membrane levels, only show poor PrPSc conversion.Citation39,67 Until to date, there are no plausible explanations for this curious and puzzling finding in this widely used PrPC-overexpressing mouse model. We suggest that shed PrP blocks PrPSc formation in the extracellular space and that increased production of shed PrP (as shown earlier for these mice)Citation38,39 may explain the phenomenon of high PrPC levels and poor PrPSc generation in Tga20 mice.Citation67 This protective effect of soluble PrP would likely be active in cis (i.e. referring to the same cell) and in trans (i.e., providing protection to neighboring or –by potential distribution via the brain interstitial flow (ISF)– even to distant cells). In fact, some studies support the view that anchorless versions of PrPC either directly inhibit the conversion to PrPSc or at least are less efficiently converted compared to membrane-anchored forms.Citation43,68–71 Fitting to this model, impaired production of shed PrPC in our ADAM10 cKO mice (possibly in connection with increased PrPC surface levels) resulted in increased PrPSc formation,Citation39 whereas overexpression of ADAM10 leads to decreased production of PrPSc.Citation34 Further support for this model might come from bank voles: this animal model has raised attention in prion research due to its high susceptibility to different sources of prions.Citation72,73 Accordingly, transgenic mice expressing bank vole PrPC are highly susceptible to prion infection. Although a detailed analysis is required, this might in part be attributed to known alterations in the amino acids sequence of bank vole PrPC in direct vicinity of the putative ADAM10 cleavage site, potentially resulting in impaired sheddingCitation74 and consequently lack of protective shed PrP along with increased membrane PrPC levels.
Prion Spreading—A Potential Role of ADAM10-Mediated Shedding?
Prion diseases and other neurodegenerative proteinopathies, such as AD or PD, have in common that pathology spreads throughout the brain following a defined pattern.Citation75–77 For acquired forms of prion diseases initiated by uptake of infectious prions (e.g., via the digestive tract) spread from the periphery to the CNS, a process termed “neuroinvasion,” is required to establish disease.Citation78,79 Although the precise mechanisms are not fully understood, it is thought that certain conformers of pathogenic PrP act as “seeds” or “nucleating particles” critical for the spread from affected to unaffected cells, tissues and brain regions.Citation75–77
Different mechanisms of spread have been suggested in the past. In vitro experiments proposed a role of direct cell-to-cell fusion and formation of tunneling nanotubes for the intercellular spread of prion seeds.Citation80 There is evidence that released membranous structures such as viral particlesCitation81,82 and especially exosomesCitation83,84 participate in dissemination of prion seeds. In addition, transsynaptic cell-to-cell transfer of pathogenic prion seeds and transport along neurites seems to be relevant.Citation79,85
Of note, all of the above mechanisms are currently thought to depend on membrane anchoring of PrPC and, indeed, the importance of membrane-bound PrP for neuroinvasion and neural spread has been described.Citation86 Nevertheless, mice expressing anchorless PrPC also spread the disease within the CNS albeit with an altered pattern.Citation87,88 These studies also discovered a contribution of the brain ISF on the dissemination of anchorless pathogenic prion seeds. Thus, we investigated in our mouse model whether proteolytically shed PrP has a similar role in the spread of prion pathology. In contrast to the disease-accelerating function that lack of PrPC shedding has, we were surprised to see that lack of ADAM10-mediated shedding of PrPC impaired spreading of prion pathology from the inoculation site to distant brain regions.Citation39 In contrast, efficient shedding in Tga20 mice was associated with efficient spread throughout the brain. This is in line with data from the mouse model expressing anchorless PrPC where extensive spread of PrPSc also to regions outside the brain has been documented.Citation89
Our finding of clearly reduced prion-associated pathology in cerebellum and brain stem of intracerebrally inoculated ADAM10 cKO mice could indicate that—in addition to the mechanisms of prion spread listed above—the protease ADAM10 contributes to the dissemination of pathology within the CNS.Citation39 This could potentially occur by releasing a specific disease-associated cluster or conformer of PrP (as discussed in the next chapter) into the extracellular space, which—via the brain ISF or other routes—is able to nucleate misfolding of PrPC molecules in distant brain areas. Since both PrPC and its sheddase are ubiquitously expressed throughout the body, this might also bear relevance for the neuroinvasion of exogenously acquired prions.
Disparity Between PrPSc Levels, Formation of Infectious Prions and Spread of Pathology
According to the protein-only hypothesis, infectious prion particles are devoid of nucleic acids and mainly –if not solely– composed of misfolded prion protein (PrPSc or PrPres).Citation90 In fact, many studies supported the concurrence of PrPSc and prion infectivity.Citation91–93 The potential need of non-PrP cofactors for the formation of prion particlesCitation94–96 has recently been questioned when infectious prions were generated from recombinant prion protein.Citation97 We were interested to study whether lack of PrP shedding in our ADAM10 cKO mice would also influence the formation of infectious prions. In view of the significantly increased PrPSc levels found in prion-infected ADAM10 cKO mice, we were surprised to find unchanged infectivity titers.Citation39 Thus, our data might indicate that amounts of PrPSc on the one hand and prion infectivity on the other hand are at least not directly congruent. In this respect our study fits to other reports showing some degree of disparity between these 2 sides of the same coin.Citation98–104 While the necessity of PrPSc for the formation of transmissible prions is largely undoubted, the exact composition and structure of these particles is still unknown.Citation105 Accordingly, while lack of ADAM10-mediated shedding in our study clearly affects PrPSc production, it does not seem to directly impact on generation of prion infectivity.Citation39 Generation of prions seems to depend on more than just formation of PrPSc and likely requires its assembly into unique structures.Citation99 A recent study described higher infectivity upon prion-challenge in mice expressing anchorless PrPC compared to wild type mice.Citation106 However, since in this study the effect was only seen in recipient mice with dramatic overexpression of PrPC, results cannot be compared to our study where physiological shedding of PrPC is only abolished in a small subset of total brain cells.Citation39
Interestingly, our study also suggested an influence of ADAM10-mediated shedding on the spread of prion-associated pathology within the brain.Citation39 In the light of unaltered infectivity titers this could indicate existence of 2 (or more) different entities with bona fide prions accounting for transmission, whereas differently composed or shaped particles might be responsible for the spread of disease throughout the CNS. How could this be explained? It has been shown that not only PrPC but likewise PrPSc can be proteolytically shed.Citation24,28,33 In contrast, experimental release by PI-PLC can only be achieved for PrPC indicating that conformational changes in PrPSc still allow for access of the protease ADAM10 but hinder phospholipase cleavage of the GPI moiety.Citation107 ADAM10-mediated release of a critical seed size of oligomeric PrPSc might –together with other mechanisms mentioned above– contribute to prion spread whereas generation of infectious prions may be independent of the protease's action. In other words, given that the exact composition and structure of the responsible entities is still unsolved, it is possible that ADAM10 influences intracerebral spread of pathology whereas it is rather not decisive or rate-limiting for the production of prions. Importantly, varying contributions of different cell types and potential cofactors in fine-tuning the production of these different forms in the context of prion diseases cannot be ruled out and deserve further investigations.
PrP Shedding and its Potential Impact on Other Neurodegenerative Proteinopathies
As described above, GPI-anchored PrPC acts as a high affinity receptor for Aβ oligomers at the neuronal membraneCitation55 and likely does so for different neurotoxic conformers associated with other neurodegenerative diseases.Citation15 Since ADAM10-mediated shedding reduces PrPC at the plasma membrane, its stimulation could offer a therapeutic option to reduce neurotoxicity. Moreover, similarly to the inhibitory effect of soluble PrPC on PrPSc formation in vivo,Citation34,39,43 recombinant or transgenic anchorless PrP binds Aβ oligomers in the extracellular space and blocks their toxic access to neurons.Citation17,108,109 In contrast to these protective effects in AD, one report showed that binding of recombinant PrP to Aβ fibrils leads to their disassembly to more toxic oligomeric forms of Aβ.Citation110 Thus, effects of anchorless or physiologically shed PrP on these and other toxic protein oligomers, such as α-synuclein, remain to be investigated in more detail.
Open Questions and Perspectives
Apart from the pathogenic aspects discussed above, it appears likely that PrPC shedding serves physiological functions. This assumption is especially supported by the high degree of conservation of this cleavage event and the rather ubiquitous expression of protease (ADAM10) and substrate (PrPC). Given that, surprisingly little is known about potential biological activities of shed PrPC. In one study, primary neurons were incubated with recombinant PrP and this led to polarization and synapse formation.Citation111 Although recombinant PrP mimics shed PrP with regard to the lack of a GPI-anchor, other structural features (such as a lack of N-glycans) do not exactly recapitulate shed PrP and, thus, findings obtained with recombinant protein cannot directly be extrapolated to its physiological correlate (shed PrPC).
So far, PrPC is one of very few GPI-anchored ADAM10 substrates identified, whereas the majority of substrates contain a transmembrane domain and are thus subject to ectodomain shedding. However, it has been shown that minor fractions of PrP are produced as cytosolic or transmembrane forms.Citation112,113 It remains to be investigated whether the latter are subject to membrane-proximate cleavage by ADAM10 and if this has physiological or pathological consequences. Interestingly, many other ADAM10 substrates in the brain have key roles in one or more of the following aspects: (i) developmental processes (e.g. NotchCitation114), (ii) cell adhesion (e.g., ephrinsCitation115 and N-cadherinCitation116), (iii) synapse integrity and function (Citation117; e.g. neuroligin-1Citation118) as well as (iV) neuroprotection (e.g., sAPPα derived from APPCitation37,119). ADAM10-mediated shedding plays a major role in regulating these aspects. Interestingly, all of these aspects have also been attributed to PrPC (for a review see refs. Citation120, 121). Despite the fact that the physiological roles played by PrPC are still not fully understood, an interesting speculation may arise from this connection: Could the fact that PrPC is a conserved substrate of the highly brain-relevant sheddase ADAM10 tell us something about its physiological functions? Moreover, could shed PrPC (as discussed here) and other truncated forms produced by physiological cleavage events (discussed elsewhere; for a review see refs. Citation18–20)—at least in part—even represent a key to understand the multitude of suggested PrPC functions? And finally, could there be even more yet overlooked cleavage events on PrP that bear physiological or pathogenic relevance ()? Future research should gain deeper insight into these aspects and hopefully provide new therapeutic strategies.
Figure 2. Important questions regarding the shedding of the prion protein. A selection is mentioned in this box. A detailed discussion can be found in the main text.

Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to apologize to all authors whose important contributions to this field of prion research could not be cited due to space limitations.
Funding
The work of the authors is supported by the Deutsche Forschungsgemeinschaft (DFG) (SFB877, project A12 (to MG and HCA), GRK1459 (to MG) and FOR885 (to MG)) and by the Werner-Otto-Stiftung, Hamburg, Germany (to HCA and LL).
REFERENCES
- Riesner D. Biochemistry and structure of PrP(C) and PrP(Sc). Br Med Bull 2003; 66:21–33; PMID:14522846; http://dx.doi.org/10.1093/bmb/66.1.21
- Biasini E, Turnbaugh JA, Unterberger U, Harris DA. Prion protein at the crossroads of physiology and disease. Trends Neurosci 2012; 35:92-103; PMID:22137337; http://dx.doi.org/10.1016/j.tins.2011.10.002
- Head MW. Human prion diseases: molecular, cellular and population biology. Neuropathology 2013; 33:221-36; PMID:23331517; http://dx.doi.org/10.1111/neup.12016
- Imran M, Mahmood S. An overview of animal prion diseases. Virol J 2011; 8:493; PMID:22044871; http://dx.doi.org/10.1186/1743-422X-8-493
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363-83; PMID:9811807; http://dx.doi.org/10.1073/pnas.95.23.13363
- Supattapone S. Prion protein conversion in vitro. J Mol Med 2004; 82(6):348-56; PMID:15014886
- Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog 2009; 5:e1000421; PMID:19436715; http://dx.doi.org/10.1371/journal.ppat.1000421
- Silva CJ, Vazquez-Fernandez E, Onisko B, Requena JR. Proteinase K and the structure of PrP: the good, the bad and the ugly. Virus Res 2015; PMID:25816779
- Harris DA, Huber MT, van Dijken P, Shyng SL, Chait BT, Wang R. Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry 1993; 32:1009-16; PMID:8093841; http://dx.doi.org/10.1021/bi00055a003
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987; 51:229-40; PMID:2444340; http://dx.doi.org/10.1016/0092-8674(87)90150-4
- Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 2010; 13:310-8; PMID:20098419; http://dx.doi.org/10.1038/nn.2483
- Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J Biol Chem 2011; 286:44234-42; PMID:22025612; http://dx.doi.org/10.1074/jbc.M111.286195
- Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 2009; 284:35973-86; PMID:19850936; http://dx.doi.org/10.1074/jbc.M109.051086
- Guillot-Sestier MV, Sunyach C, Ferreira ST, Marzolo MP, Bauer C, Thevenet A, Checler F. alpha-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid beta (Abeta)-associated cell death. J Biol Chem 2012; 287:5021-32; PMID:22184125; http://dx.doi.org/10.1074/jbc.M111.323626
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Muller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, et al. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J 2011; 30:2057-70; PMID:21441896; http://dx.doi.org/10.1038/emboj.2011.86
- Beland M, Motard J, Barbarin A, Roucou X. PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J Neurosci 2012; 32:13255-63; PMID:22993441; http://dx.doi.org/10.1523/JNEUROSCI.2236-12.2012
- Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, et al. An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J Biol Chem 2013; 288:7857-66; PMID:23362282; http://dx.doi.org/10.1074/jbc.M112.423954
- Liang J, Kong Q. alpha-Cleavage of cellular prion protein. Prion 2012; 6:453-60; PMID:23052041; http://dx.doi.org/10.4161/pri.22511
- Altmeppen HC, Puig B, Dohler F, Thurm DK, Falker C, Krasemann S, Glatzel M. Proteolytic processing of the prion protein in health and disease. Am J Neurodegener Dis 2012; 1:15-31; PMID:23383379
- Altmeppen HC, Prox J, Puig B, Dohler F, Falker C, Krasemann S, Glatzel M. Roles of endoproteolytic alpha-cleavage and shedding of the prion protein in neurodegeneration. FEBS J 2013; 280:4338-47; PMID:23413979; http://dx.doi.org/10.1111/febs.12196
- Tagliavini F, Prelli F, Porro M, Salmona M, Bugiani O, Frangione B. A soluble form of prion protein in human cerebrospinal fluid: implications for prion-related encephalopathies. Biochem Biophys Res Commun 1992; 184:1398-404; PMID:1375461; http://dx.doi.org/10.1016/S0006-291X(05)80038-5
- Perini F, Vidal R, Ghetti B, Tagliavini F, Frangione B, Prelli F. PrP27-30 is a normal soluble prion protein fragment released by human platelets. Biochem Biophys Res Commun 1996; 223:572-7; PMID:8687437; http://dx.doi.org/10.1006/bbrc.1996.0936
- MacGregor I, Hope J, Barnard G, Kirby L, Drummond O, Pepper D, Hornsey V, Barclay R, Bessos H, Turner M, et al. Application of a time-resolved fluoroimmunoassay for the analysis of normal prion protein in human blood and its components. Vox Sang 1999; 77:88-96; PMID:10516553; http://dx.doi.org/10.1046/j.1423-0410.1999.7720088.x
- Borchelt DR, Rogers M, Stahl N, Telling G, Prusiner SB. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology 1993; 3:319-29; PMID:7691278; http://dx.doi.org/10.1093/glycob/3.4.319
- Parizek P, Roeckl C, Weber J, Flechsig E, Aguzzi A, Raeber AJ. Similar turnover and shedding of the cellular prion protein in primary lymphoid and neuronal cells. J Biol Chem 2001; 276:44627-32; PMID:11571302; http://dx.doi.org/10.1074/jbc.M107458200
- Zhao H, Klingeborn M, Simonsson M, Linne T. Proteolytic cleavage and shedding of the bovine prion protein in two cell culture systems. Virus Res 2006; 115:43-55; PMID:16140411; http://dx.doi.org/10.1016/j.virusres.2005.07.004
- Parkin ET, Watt NT, Turner AJ, Hooper NM. Dual mechanisms for shedding of the cellular prion protein. J Biol Chem 2004; 279:11170-8; PMID:14711812; http://dx.doi.org/10.1074/jbc.M312105200
- Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, Hooper NM. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem 2009; 284:22590-600; PMID:19564338; http://dx.doi.org/10.1074/jbc.M109.032599
- McDonald AJ, Dibble JP, Evans EG, Millhauser GL. A new paradigm for enzymatic control of alpha-cleavage and beta-cleavage of the prion protein. J Biol Chem 2014; 289:803-13; PMID:24247244; http://dx.doi.org/10.1074/jbc.M113.502351
- Cisse MA, Sunyach C, Lefranc-Jullien S, Postina R, Vincent B, Checler F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J Biol Chem 2005; 280:40624-31; PMID:16236709; http://dx.doi.org/10.1074/jbc.M506069200
- Tousseyn T, Thathiah A, Jorissen E, Raemaekers T, Konietzko U, Reiss K, Maes E, Snellinx A, Serneels L, Nyabi O, et al. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J Biol Chem 2009; 284:11738-47; PMID:19213735; http://dx.doi.org/10.1074/jbc.M805894200
- Moss ML, Powell G, Miller MA, Edwards L, Qi B, Sang QX, De Strooper B, Tesseur I, Lichtenthaler SF, Taverna M, et al. ADAM9 inhibition increases membrane activity of ADAM10 and controls alpha-secretase processing of amyloid precursor protein. J Biol Chem 2011; 286:40443-51; PMID:21956108; http://dx.doi.org/10.1074/jbc.M111.280495
- Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990; 29:8879-84; PMID:1980209; http://dx.doi.org/10.1021/bi00490a001
- Endres K, Mitteregger G, Kojro E, Kretzschmar H, Fahrenholz F. Influence of ADAM10 on prion protein processing and scrapie infectiosity in vivo. Neurobiol Dis 2009; 36:233-41; PMID:19632330; http://dx.doi.org/10.1016/j.nbd.2009.07.015
- Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lübke T, Lena Illert A, von Figura K, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet 2002; 11:2615-24; http://dx.doi.org/10.1093/hmg/11.21.2615
- Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci 2010; 30:4833-44; http://dx.doi.org/10.1523/JNEUROSCI.5221-09.2010
- Prox J, Bernreuther C, Altmeppen H, Grendel J, Glatzel M, D'Hooge R, Stroobants S, Ahmed T, Balschun D, Willem M, et al. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J Neurosci 2013; 33:12915-28, 28a; PMID:23926248; http://dx.doi.org/10.1523/JNEUROSCI.5910-12.2013
- Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, Jorissen E, Petrowitz B, Bartsch U, De Strooper B, et al. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener 2011; 6:36; PMID:21619641; http://dx.doi.org/10.1186/1750-1326-6-36
- Altmeppen HC, Prox J, Krasemann S, Puig B, Kruszewski K, Dohler F, Bernreuther C, Hoxha A, Linsenmeier L, Sikorska B, et al. The sheddase ADAM10 is a potent modulator of prion disease. eLife 2015; 4; PMID:25654651; doi:10.7554/eLife.04260
- McNally KL, Ward AE, Priola SA. Cells expressing anchorless prion protein are resistant to scrapie infection. J Virol 2009; 83:4469-75; PMID:19225008; http://dx.doi.org/10.1128/JVI.02412-08
- Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A 2001; 98:9295-9; PMID:11470893; http://dx.doi.org/10.1073/pnas.151242598
- Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem 1991; 266:18217-23; PMID:1680859
- Meier P, Genoud N, Prinz M, Maissen M, Rulicke T, Zurbriggen A, Raeber AJ, Aguzzi A. Soluble dimeric prion protein binds PrP(Sc) in vivo and antagonizes prion disease. Cell 2003; 113:49-60; PMID:12679034; http://dx.doi.org/10.1016/S0092-8674(03)00201-0
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435-9; PMID:15933194; http://dx.doi.org/10.1126/science.1110837
- Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog 2010; 6:e1000800; PMID:20221436; http://dx.doi.org/10.1371/journal.ppat.1000800
- Stohr J, Watts JC, Legname G, Oehler A, Lemus A, Nguyen HO, Sussman J, Wille H, DeArmond SJ, Prusiner SB, et al. Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci U S A 2011; 108:21223-8; PMID:22160704; http://dx.doi.org/10.1073/pnas.1117827108
- Büeler HR, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339-47; PMID:8100741; http://dx.doi.org/10.1016/0092-8674(93)90360-3
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996; 379:339-43; PMID:8552188; http://dx.doi.org/10.1038/379339a0
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003; 302:871-4; PMID:14593181; http://dx.doi.org/10.1126/science.1090187
- Resenberger UK, Winklhofer KF, Tatzelt J. Neuroprotective and neurotoxic signaling by the prion protein. Top Curr Chem 2011; 305:101-19; PMID:21598098; http://dx.doi.org/10.1007/128_2011_160
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540-2; PMID:21350487; http://dx.doi.org/10.1038/nature09768
- Radford HE, Mallucci GR. The role of GPI-anchored PrP C in mediating the neurotoxic effect of scrapie prions in neurons. Curr Issues Mol Biol 2010; 12:119-27; PMID:19767655
- Puig B, Altmeppen H, Glatzel M. The GPI-anchoring of PrP: Implications in sorting and pathogenesis. Prion 2014; 8:11-8; PMID:24509692
- Priola SA, McNally KL. The role of the prion protein membrane anchor in prion infection. Prion 2009; 3:134-8; PMID:19786843; http://dx.doi.org/10.4161/pri.3.3.9771
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128-32; PMID:19242475; http://dx.doi.org/10.1038/nature07761
- Turnbaugh JA, Unterberger U, Saa P, Massignan T, Fluharty BR, Bowman FP, Miller MB, Supattapone S, Biasini E, Harris DA. The N-terminal, polybasic region of PrP(C) dictates the efficiency of prion propagation by binding to PrP(Sc). J Neurosci 2012; 32:8817-30; PMID:22745483; http://dx.doi.org/10.1523/JNEUROSCI.1103-12.2012
- Dohler F, Sepulveda-Falla D, Krasemann S, Altmeppen H, Schluter H, Hildebrand D, Zerr I, Matschke J, Glatzel M. High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer's disease. Brain 2014; 137:873-86; PMID:24519981; http://dx.doi.org/10.1093/brain/awt375
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 2012; 15:1227-35; PMID:22820466; http://dx.doi.org/10.1038/nn.3178
- Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesné SE. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J Neurosci 2012; 32:16857-71a; PMID:23175838; http://dx.doi.org/10.1523/JNEUROSCI.1858-12.2012
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289:1925-8; PMID:10988071; http://dx.doi.org/10.1126/science.289.5486.1925
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 2005; 280:17294-300; PMID:15722360; http://dx.doi.org/10.1074/jbc.M500997200
- Kagan BL. Membrane pores in the pathogenesis of neurodegenerative disease. Prog Mol Biol Transl Sci 2012; 107:295-325; PMID:22482454; http://dx.doi.org/10.1016/B978-0-12-385883-2.00001-1
- Solomon IH, Biasini E, Harris DA. Ion channels induced by the prion protein: mediators of neurotoxicity. Prion 2012; 6:40-5; PMID:22453177; http://dx.doi.org/10.4161/pri.6.1.18627
- Sonati T, Reimann RR, Falsig J, Baral PK, O'Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013; PMID:23903654
- Goold R, McKinnon C, Rabbanian S, Collinge J, Schiavo G, Tabrizi SJ. Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J Cell Sci 2013; 126:3552-62; PMID:23813960; http://dx.doi.org/10.1242/jcs.120477
- Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, Clarke AR, Schiavo G, Jat P, Collinge J, et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun 2011; 2:281; PMID:21505437; http://dx.doi.org/10.1038/ncomms1282
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 1996; 15:1255-64; PMID:8635458
- Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 1995; 129:121-32; PMID:7698979; http://dx.doi.org/10.1083/jcb.129.1.121
- Yuan J, Zhan YA, Abskharon R, Xiao X, Martinez MC, Zhou X, Kneale G, Mikol J, Lehmann S, Surewicz WK, et al. Recombinant human prion protein inhibits prion propagation in vitro. Sci Rep 2013; 3:2911; PMID:24105336
- Marella M, Lehmann S, Grassi J, Chabry J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J Biol Chem 2002; 277:25457-64; PMID:11994310; http://dx.doi.org/10.1074/jbc.M203248200
- Kim JI, Surewicz K, Gambetti P, Surewicz WK. The role of glycophosphatidylinositol anchor in the amplification of the scrapie isoform of prion protein in vitro. FEBS Lett 2009; 583:3671-5; PMID:19854187; http://dx.doi.org/10.1016/j.febslet.2009.10.049
- Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell'Omo G, Cartoni C, Ingrosso L, Boyle A, Galeno R, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2006; 2:e12; PMID:16518470; http://dx.doi.org/10.1371/journal.ppat.0020012
- Di Bari MA, Nonno R, Castilla J, D'Agostino C, Pirisinu L, Riccardi G, Conte M, Richt J, Kunkle R, Langeveld J, et al. Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog 2013; 9:e1003219; PMID:23505374; http://dx.doi.org/10.1371/journal.ppat.1003219
- Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 2014; 10:e1003990; PMID:24699458; http://dx.doi.org/10.1371/journal.ppat.1003990
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009; 64:783-90; PMID:20064386; http://dx.doi.org/10.1016/j.neuron.2009.12.016
- Costanzo M, Zurzolo C. The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem J 2013; 452:1-17; PMID:23614720; http://dx.doi.org/10.1042/BJ20130484
- Walker LC, Jucker M. Neurodegenerative Diseases: Expanding the Prion Concept. Annu Rev Neurosci 2015; PMID:25840008
- Glatzel M, Aguzzi A. PrP(C) expression in the peripheral nervous system is a determinant of prion neuroinvasion. J Gen Virol 2000; 81:2813-21; PMID:11038396
- Glatzel M, Heppner FL, Albers KM, Aguzzi A. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 2001; 31:25-34; PMID:11498048; http://dx.doi.org/10.1016/S0896-6273(01)00331-2
- Gousset K, Zurzolo C. Tunnelling nanotubes: a highway for prion spreading? Prion 2009; 3:94-8; PMID:19471116; http://dx.doi.org/10.4161/pri.3.2.8917
- Leblanc P, Alais S, Porto-Carreiro I, Lehmann S, Grassi J, Raposo G, Darlix JL. Retrovirus infection strongly enhances scrapie infectivity release in cell culture. Embo J 2006; 25:2674-85; PMID:16724107; http://dx.doi.org/10.1038/sj.emboj.7601162
- Krasemann S, Neumann M, Luepke JP, Grashorn J, Wurr S, Stocking C, Glatzel M. Persistent retroviral infection with MoMuLV influences neuropathological signature and phenotype of prion disease. Acta Neuropathol 2012; 124:111-26; PMID:22271154; http://dx.doi.org/10.1007/s00401-012-0944-1
- Alais S, Simoes S, Baas D, Lehmann S, Raposo G, Darlix JL, Leblanc P. Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol Cell 2008; 100:603-15; PMID:18422484; http://dx.doi.org/10.1042/BC20080025
- Coleman BM, Hill AF. Extracellular vesicles - Their role in the packaging and spread of misfolded proteins associated with neurodegenerative diseases. Semin Cell Dev Biol 2015; PMID:25704308
- Shearin H, Bessen RA. Axonal and transynaptic spread of prions. J Virol 2014; 88(15):8640-55; PMID:24850738
- Klingeborn M, Race B, Meade-White KD, Rosenke R, Striebel JF, Chesebro B. Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol 2011; 85:1484-94; PMID:21123371; http://dx.doi.org/10.1128/JVI.02167-10
- Rangel A, Race B, Klingeborn M, Striebel J, Chesebro B. Unusual cerebral vascular prion protein amyloid distribution in scrapie-infected transgenic mice expressing anchorless prion protein. Acta Neuropathol Commun 2013; 1:25; PMID:24252347; http://dx.doi.org/10.1186/2051-5960-1-25
- Rangel A, Race B, Phillips K, Striebel J, Kurtz N, Chesebro B. Distinct patterns of spread of prion infection in brains of mice expressing anchorless or anchored forms of prion protein. Acta Neuropathol Commun 2014; 2:8; PMID:24447368; http://dx.doi.org/10.1186/2051-5960-2-8
- Trifilo MJ, Yajima T, Gu Y, Dalton N, Peterson KL, Race RE, Meade-White K, Portis JL, Masliah E, Knowlton KU, et al. Prion-induced amyloid heart disease with high blood infectivity in transgenic mice. Science 2006; 313:94-7; PMID:16825571; http://dx.doi.org/10.1126/science.1128635
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136-44; PMID:6801762; http://dx.doi.org/10.1126/science.6801762
- Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell 2005; 121:195-206; PMID:15851027; http://dx.doi.org/10.1016/j.cell.2005.02.011
- Weber P, Giese A, Piening N, Mitteregger G, Thomzig A, Beekes M, Kretzschmar HA. Cell-free formation of misfolded prion protein with authentic prion infectivity. Proc Natl Acad Sci U S A 2006; 103:15818-23; PMID:17030802; http://dx.doi.org/10.1073/pnas.0605608103
- Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A 2007; 104:9741-6; PMID:17535913; http://dx.doi.org/10.1073/pnas.0702662104
- Caughey B, Baron GS. Prions and their partners in crime. Nature 2006; 443:803-10; PMID:17051207; http://dx.doi.org/10.1038/nature05294
- Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A 2012; 109:E1938-46; PMID:22711839; http://dx.doi.org/10.1073/pnas.1206999109
- Supattapone S. Elucidating the role of cofactors in mammalian prion propagation. Prion 2014; 8:100-5; PMID:24365977; http://dx.doi.org/10.4161/pri.27501
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132-5; PMID:20110469; http://dx.doi.org/10.1126/science.1183748
- Lasmezas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, Fournier JG, Hauw JJ, Rossier J, Dormont D. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 1997; 275:402-5; PMID:8994041; http://dx.doi.org/10.1126/science.275.5298.402
- Shaked GM, Fridlander G, Meiner Z, Taraboulos A, Gabizon R. Protease-resistant and detergent-insoluble prion protein is not necessarily associated with prion infectivity. J Biol Chem 1999; 274:17981-6; PMID:10364247; http://dx.doi.org/10.1074/jbc.274.25.17981
- Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, Manson JC. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J Biol Chem 2007; 282:35878-86; PMID:17923484; http://dx.doi.org/10.1074/jbc.M704329200
- Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci U S A 2007; 104:4712-7; PMID:17360589; http://dx.doi.org/10.1073/pnas.0609241104
- Klingeborn M, Race B, Meade-White KD, Chesebro B. Lower specific infectivity of protease-resistant prion protein generated in cell-free reactions. Proc Natl Acad Sci U S A 2011; 108:E1244-53; PMID:22065744; http://dx.doi.org/10.1073/pnas.1111255108
- Lewis V, Haigh CL, Masters CL, Hill AF, Lawson VA, Collins SJ. Prion subcellular fractionation reveals infectivity spectrum, with a high titre-low PrPres level disparity. Mol Neurodegener 2012; 7:18; PMID:22534096; http://dx.doi.org/10.1186/1750-1326-7-18
- Krasemann S, Neumann M, Szalay B, Stocking C, Glatzel M. Protease-sensitive prion species in neoplastic spleens of prion-infected mice with uncoupling of PrP(Sc) and prion infectivity. J Gen Virol 2013; 94:453-63; PMID:23136363; http://dx.doi.org/10.1099/vir.0.045922-0
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature 2005; 437:257-61; PMID:16148934; http://dx.doi.org/10.1038/nature03989
- Race B, Phillips K, Meade-White K, Striebel J, Chesebro B. Increased infectivity of anchorless mouse scrapie prions in transgenic mice overexpressing human prion protein. J Virol 2015
- Stahl N, Borchelt DR, Prusiner SB. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry 1990; 29:5405-12; PMID:1974460; http://dx.doi.org/10.1021/bi00474a028
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2010; 2:306-14; PMID:20665634; http://dx.doi.org/10.1002/emmm.201000082
- Nieznanski K, Choi JK, Chen S, Surewicz K, Surewicz WK. Soluble Prion Protein Inhibits Amyloid-beta (Abeta) Fibrillization and Toxicity. J Biol Chem 2012; 287:33104-8; PMID:22915585; http://dx.doi.org/10.1074/jbc.C112.400614
- Younan ND, Sarell CJ, Davies P, Brown DR, Viles JH. The cellular prion protein traps Alzheimer's Abeta in an oligomeric form and disassembles amyloid fibers. FASEB J 2013; 27:1847-58; PMID:23335053; http://dx.doi.org/10.1096/fj.12-222588
- Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem 2005; 95:1373-86; PMID:16313516; http://dx.doi.org/10.1111/j.1471-4159.2005.03469.x
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998; 279:827-34; PMID:9452375; http://dx.doi.org/10.1126/science.279.5352.827
- Emerman AB, Zhang ZR, Chakrabarti O, Hegde RS. Compartment-restricted biotinylation reveals novel features of prion protein metabolism in vivo. Mol Biol Cell 2010; 21:4325-37; PMID:20980618; http://dx.doi.org/10.1091/mbc.E10-09-0742
- Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development 2012; 139:3693-709; PMID:22991436; http://dx.doi.org/10.1242/dev.076398
- Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell 2005; 123:291-304; PMID:16239146; http://dx.doi.org/10.1016/j.cell.2005.08.014
- Reiss K, Maretzky T, Ludwig A, Tousseyn T, de Strooper B, Hartmann D, Saftig P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. Embo J 2005; 24:742-52; PMID:15692570; http://dx.doi.org/10.1038/sj.emboj.7600548
- Musardo S, Marcello E, Gardoni F, Di Luca M. ADAM10 in synaptic physiology and pathology. Neurodegener Dis 2014; 13:72-4; PMID:24008925; http://dx.doi.org/10.1159/000354233
- Suzuki K, Hayashi Y, Nakahara S, Kumazaki H, Prox J, Horiuchi K, Zeng M, Tanimura S, Nishiyama Y, Osawa S, et al. Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 2012; 76:410-22; PMID:23083742; http://dx.doi.org/10.1016/j.neuron.2012.10.003
- Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J 2010; 29:3020-32; PMID:20676056; http://dx.doi.org/10.1038/emboj.2010.167
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci 2008; 31:439-77; PMID:18558863; http://dx.doi.org/10.1146/annurev.neuro.31.060407.125620
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev 2008; 88:673-728; PMID:18391177; http://dx.doi.org/10.1152/physrev.00007.2007