
ABSTRACT
Tauopathies are a family of neurodegenerative diseases in which fibrils of human hyperphosphorylated tau (P-tau) are believed to cause neuropathology. In Alzheimer disease, P-tau associates with A-beta amyloid and contributes to disease pathogenesis. In familial human prion diseases and variant CJD, P-tau often co-associates with prion protein amyloid, and might also accelerate disease progression. To test this latter possibility, here we compared progression of amyloid prion disease in vivo after scrapie infection of mice with and without expression of human tau. The mice used expressed both anchorless prion protein (PrP) and membrane-anchored PrP, that generate disease associated amyloid and non-amyloid PrP (PrPSc) after scrapie infection. Human P-tau induced by scrapie infection was only rarely associated with non-amyloid PrPSc, but abundant human P-tau was detected at extracellular, perivascular and axonal deposits associated with amyloid PrPSc. This pathology was quite similar to that seen in familial prion diseases. However, association of human and mouse P-tau with amyloid PrPSc did not diminish survival time following prion infection in these mice. By analogy, human P-tau may not affect prion disease progression in humans. Alternatively, these results might be due to other factors, including rapidity of disease, blocking effects by mouse tau, or low toxicity of human P-tau in this model.
INTRODUCTION
Tauopathies are a heterogeneous group of human neurodegenerative disorders where the neuronal brain protein, tau, becomes hyperphosphorylated, and detaches from microtubules to form potentially pathogenic oligomers, aggregates and neurofibrillary tangles (NFT) (for review see refs. Citation1, Citation2). Tauopathies include frontotemporal dementia with parkinsonism linked to chromosome 17, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. In all these diseases, deposits of aggregated phosphorylated tau (P-tau) in brain are associated with progressive motor and behavioral abnormalities. In Alzheimer disease (AD), tau NFT and amyloid-β plaques are the 2 main pathological lesions observed, and they are often co-localized.Citation3 Progression of dementia in AD correlates well with accumulation of tau NFT suggesting an important role in pathogenesis.Citation4 P-tau deposits have also been detected in association with amyloid in familial British dementia, and thus P-tau may also play a role in pathogenesis of this disease.Citation5
Abnormal deposits of P-tau have been detected in familial human prion diseases, such as GSS with mutations F198S, Q217R and Y145Stop.Citation6 Similar P-tau deposits were also noted in infectious human prion diseases, such as vCJD in humansCitation7 and BSE in non-human primates (lemurs).Citation8 In all these situations, P-tau deposits were closely associated with prion protein amyloid, composed of the disease-associated protease-resistant form of prion protein (PrPSc) organized as amyloid fibrils. The presence of P-tau deposits in non-amyloid prion diseases is less common, but has recently been shown in squirrel monkeys infected with BSE Citation9 and in some, but not all human sCJD cases.Citation5,10
Genetic manipulation of the expression of mouse or human tau in mice has been used to test the effect of tau expression on survival time after experimental prion infection. Lawson and co-authors used the M1000 strain of mouse-adapted human prions to infect control mice versus mice deleted for expression of mouse tau.Citation11 In mice expressing mouse tau, altered forms of P-tau were detected by immunoblot, and mouse P-tau cytoplasmic staining was observed in a subpopulation of neurons in hippocampus and thalamus. However, no alteration in survival curves was seen by comparing mice with or without expression of mouse tau. It is possible that this result was due to lack of formation of pathogenic fibrils by mouse tau in vivo.Citation12,13 In another study, after intracerebral infection with mouse prions (RML strain), mice expressing a 4-repeat (4R) human tau transgene showed accumulation of human P-tau, but there was no difference in onset of terminal scrapie compare to non-transgenic control mice.Citation14 However, this model using the RML scrapie strain generated exclusively the diffuse non-amyloid form of PrPSc, whereas in previous reports deposits of P-tau aggregates were mostly associated with the amyloid form of PrPSc.Citation6,7
In the present paper, we tested the role of human tau in prion disease using a mouse prion disease model that forms both PrPSc amyloid plaques and non-amyloid diffuse PrPSc deposits. Mice hemizygous for a transgene expressing human tau in neuronsCitation15 were bred with mice homozygous for a transgene expressing GPI anchorless PrP (tg44 mice), which develop extensive amyloid plaque pathology during prion infectionCitation16. The resulting F1 mice expressed both the anchorless and GPI-anchored forms of mouse PrP, and were previously shown to generate both the amyloid and non-amyloid forms of PrPSc during scrapie infection.Citation17 In the present experiments, the human tau transgene was present in 50% of our F1 mice, and thus these mice were either hemizygous for the human transgene, (huTau+) or null for the human tau transgene (huTau−). All mice were hemizygous for mouse tau, GPI-anchored PrP, and GPI-anchorless PrP. Groups of mice derived from F1 littermates were inoculated intracerebrally with mouse scrapie, and comparisons were made between huTau+ and huTau− mice. Several variables were evaluated, including survival curves, deposition of P-tau and association of P-tau with various forms of PrPSc.
RESULTS
Scrapie Survival Analysis
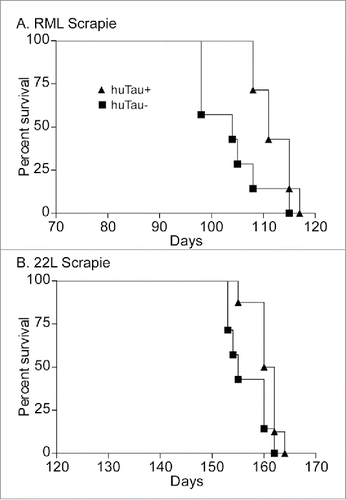
To determine if human tau contributed to overall prion disease pathogenesis we intracerebrally inoculated both huTau+ and huTau− mice with either 22L or RML rodent-adapted scrapie strains. Groups of mice were age and gender matched, and all inoculations for a given scrapie strain were completed on the same day with the same dilution of inoculum. Observers were blinded to the mouse genotypes, and mice were euthanized at an advanced stage of clinical disease. Clinical signs did not differ between huTau+ and huTau− mice, and were consistent with clinical signs previously observed in mouse models with anchored PrP.Citation16 Overexpression of human tau did not increase disease tempo with either scrapie strain 22L or RML. With both scrapie strains, the presence of human tau was mildly beneficial to survival times with an increase in average survival time of huTau+ mice of 8 d for the RML scrapie experiment () and 4 d for the 22L experiment (). Thus in this prion protein amyloid plaque model, human tau did not accelerate disease progression.
FIGURE 1. Survival curve analysis of scrapie-infected huTau− (solid squares) and huTau+ (solid triangles) mice. (A) RML scrapie-infected mice. Log-rank (Mantel-Cox) analysis of the curves gave a P value = 0.0242. (B) 22L scrapie-infected mice. Log-rank analysis of the curves gave a P value = 0.0457. N = 8 for all groups.
Detection of PrPSc by Immunoblot
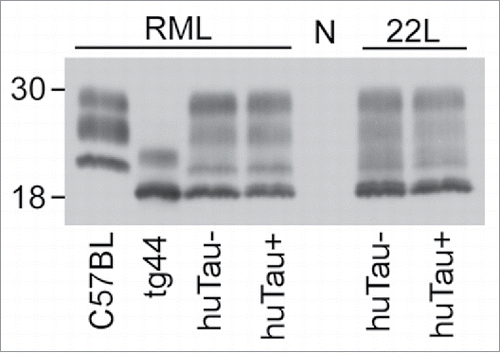
To test if expression of human tau would alter the amount or biochemical properties of PrPSc accumulation in the brain of scrapie-infected mice, we performed western immunoblots using the anti-PrP antibody D13. Brains from mice that had clinical scrapie were homogenized, subjected to proteinase K digestion (50 µg/ml) and analyzed by western blot. The banding pattern and intensity did not vary between huTau− and huTau+ mice infected with either RML or 22L scrapie, () suggesting that human tau expression did not affect PrPSc accumulation or clearance. After scrapie infection, a unique 4 band pattern was seen as a result of the combined expression of both GPI-anchored and anchorless PrP expression in this model. The lowest band corresponded to the GPI-anchorless PrPSc.
FIGURE 2. Immunoblot of PrPSc from brains of uninfected and scrapie-infected huTau− and huTau+ mice. All brain tissue on this immunoblot was digested with proteinase K (PK) prior to analysis. The presence (+) or absence (−) of the huTau transgene for each sample is shown across the bottom of the immunoblot. The strain of scrapie (RML or 22L) for each sample is shown above the immunoblot. All lanes were loaded with 4 µl freshly boiled samples as described in the methods (0.24 mg of tissue prior to PK digestion). N represents a normal, uninfected, huTau+ control mouse brain that shows complete PK digestion of PrPsen. The first 2 lanes show control RML-infected C57BL and tg44 mice respectively. For the tg44 mouse the lowest band was the unglycosylated form of tg44 anchorless PrPres, which migrated similarly to the lowest band in the infected F1 mice. The immunoblot was probed with the anti-PrP antibody D13.
Detection of P-Tau and Total Tau by Immunoblot
In order to determine the influence of scrapie infection on tau phosphorylation, brain samples were screened by immunoblot with various anti-tau antibodies. There are over 70 phosphorylation sites known in tau, and here we studied 3 sites, pS202, pS396 and pS404, that have been examined previously in prion diseases.Citation11,18-20
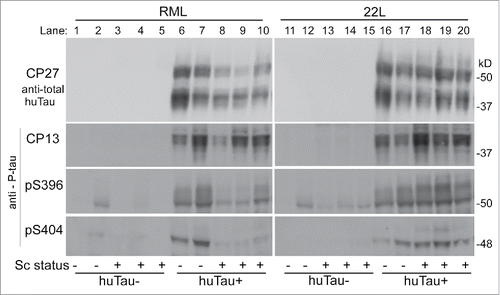
In our experiments, the 22L-infected huTau+ mice showed a slight increase in phosphorylation at S202 (antibody CP13) vs. uninfected mice (), whereas this change was not convincing in RML-infected mice. In contrast, using antibodies specific for P-tau sites S396 and S404, we observed a decrease in phosphorylation in scrapie-infected mice compared to uninfected huTau+ mice after RML-infection, but not after 22L-infection (). Thus tau phosphorylation appeared to show scrapie-related alterations, but the 2 scrapie strains tested differed in the details. Compared to human P-tau, mouse P-tau was undetectable or very low in infected or uninfected huTau− mice ().
FIGURE 3. Immunoblot of brain tissue from uninfected and infected (RML and 22L) huTau− and huTau+ mice developed with 4 different anti-tau antibodies. Antibodies are indicated to the left of each row. The first row was probed with anti-total human tau antibody, CP27, and lower 3 rows were probed with antibodies specific for P-tau phosphorylation sites pS202 (CP13), pS396, and pS404 respectively. Approximate molecular weights are shown on the right of each row.
Detection of PrPSc and P-Tau by Immunohistochemistry
To visualize human tau and determine if there was any association of human tau with PrPSc in our model, we performed immunohistochemistry (IHC) for both PrPSc and P-tau. For each experimental group, 4-6 brains were sectioned and stained. Sagittal brain sections derived from scrapie-infected mice showed widespread deposition of both PrPSc amyloid plaques and non-amyloid, diffuse PrPSc (). No differences in PrPSc distribution were noted between huTau+ and huTau− scrapie-infected mice. To confirm the amyloid nature of the PrPSc we used thioflavin S (green) to stain amyloid. In , a PrPSc amyloid plaque is surrounded by thioflavin S-negative brain tissue. In , the same amyloid plaque (yellow arrow) was also stained for PrPSc (red), while non-amyloid PrPSc could be seen in the nearby brain parenchyma, corresponding to the thioflavin S-negative areas. Often the non-amyloid PrPSc was in a pericellular or perinuclear location (black arrowheads).
FIGURE 4. Immunohistochemical analysis of PrPSc deposition and association with human tau in transgenic mouse brains. All brain tissues from scrapie-infected mice were obtained at the time of clinical disease. (A-G) Representative sections from a RML scrapie-infected huTau+ mouse and (H-J) 22L scrapie-infected huTau+ mouse are shown. (A) Whole brain sagittal section demonstrating widespread PrPSc staining with D13 (red) and boxes depicting areas enlarged in D-F. (B, C) PrPSc plaque stained with D13 and Thioflavin S respectively. (D-J) Dual staining of PrPSc with D13 (red) and P-tau using anti-P-tau antibody CP13 (brown). A clear association of P-tau aggregates can be seen around PrPSc plaques (yellow arrows) in several regions of the brain including (D) frontal dorsal cortex, (E) hypothalamus, (F,G,H) colliculus, and (I,J) dorsal cortex. Non-amyloid PrPSc (black arrowheads) rarely had P-tau staining nearby. Scrapie-associated vacuoles (blue arrowheads) could be seen in some brain regions. (K) Minimal P-tau staining by CP13 in cortex of 22L scrapie-infected huTau− mouse. (L) Absence of D13 and CP13 staining in an uninfected huTau+ mouse. (M) PrPSc amyloid plaque from a huTau+ 22L scrapie-infected mouse stained with CP13. Note, without D13 staining the localization of P-tau in ring-like structures (pink arrows) is more obvious. The scale bar in each panel is 50 µm.
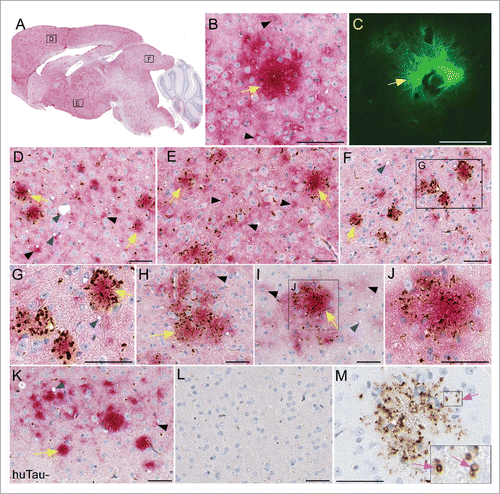
To determine whether P-tau was associated with PrPSc deposits in brain, we co-stained brain tissues with anti-P-tau antibody CP13 (brown) and anti-PrP antibody D13 (red). P-tau specific staining with CP13 showed a clear association of aggregated tau deposition around and within structures resembling PrPSc plaques in both RML () and 22L () scrapie-infected huTau+ mice. P-tau association with non-amyloid PrPSc in huTau+ () was rarely seen in our experimental mice. Interestingly, in scrapie-infected huTau− mice, CP13 staining for P-tau was weak to absent () suggesting that the P-tau staining observed with CP13 was due to human tau and not mouse tau. No PrPSc or P-tau staining was detected in uninfected huTau+ control mice () demonstrating the P-tau deposits observed were associated with scrapie infection and were not an age-related change.
In the dual-stained slides, the detailed staining characteristics of the P-tau were unclear due to the overlap with the intense red staining from the PrPSc. Therefore, slides stained with CP13 alone were examined, and here some P-tau staining appeared to localize in small ring-like structures consistent with dilated axons or capillaries (), which were quite similar to P-tau staining in human and mouse vCJD.Citation7 Other P-tau aggregates were more irregular and appeared to be extracellular sites or perhaps associated with glial cells surrounding the plaques. Dual-staining for P-tau and the glial cell markers, GFAP and Iba1, confirmed that both glial cell types were in very close association with plaques, but did not co-localize with P-tau, suggesting that glia were not harboring the P-tau detected here.
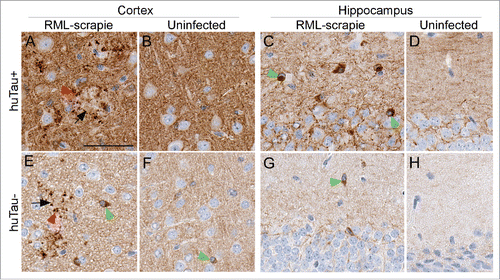
To confirm the tau staining results obtained with CP13, we tested 4 additional anti-tau antibodies by IHC. Similar plaque-associated P-tau was confirmed using 2 anti-P-tau antibodies, pS396 () and pS404 and 2 antibodies for total tau, E178 and CP27. The overall level of staining in brain tissues was also in agreement with the immunoblot data () that showed huTau+ mice had more tau staining than huTau− mice (compare amount of brown stain in ). In addition to the plaque-associated P-tau, anti-pS396 showed strong cytoplasmic staining in many large cells that appeared to be neurons, especially in the hippocampus of huTau+ mice (). Interestingly, the huTau− mice that expressed only mouse tau also had intracellular P-tau staining detectable by anti-pS396 () similar to intracellular mouse P-tau reported by others.Citation11 Only an occasional pS396- positive cell could be found in the cortex and hippocampus of uninfected huTau+ or huTau− mice ().
FIGURE 5. Immunohistochemical analysis of pS396 tau in huTau+ and huTau− mice. All brain tissues from scrapie-infected mice were obtained at the time of clinical disease. Brain region and infection status are shown across the top of the figure. The top row shows huTau+ mice, the bottom row shows huTau− mice. Black arrows indicate examples of PrPSc plaques, red arrowheads show plaque-associated P-tau, and green arrowheads show intracellular P-tau staining. The scale bar in A is 50 µm and applies to all panels.
DISCUSSION
In tauopathies and in AD there is strong evidence from human studies that phosphorylated mutant or normal tau protein contributes to neuropathogenesis.Citation2 This conclusion is supported directly by studies using transgenic and/or knockout mice showing reduced disease in the absence of tau expression or increased disease with co-expression of mutant human tau and mutant A-beta.Citation21-23 In the case of prion diseases, deposition of P-tau has been detected in GSS and vCJD brain at late clinical times, indicating a correlation of P-tau deposition with neuropathogenesis. The presence of amyloid PrPSc may be important for this effect, as P-tau deposits have only rarely been associated with non-amyloid PrPSc typical of mouse scrapie and human sCJD.Citation5,9,10 Studies using transgenic or knockout mice provide a means of directly testing whether the P-tau deposits seen at the end stage of prion disease act to reduce survival time. In two earlier reports, neither deletion of mouse tau nor expression of 4R human tau altered survival after infection with mouse-adapted prion agents.Citation11,14 However, in both of these studies the PrPSc deposited was non-amyloid PrPSc, which might be responsible for this result. Therefore, in the current experiments we used mice which generated both non-amyloid and amyloid PrPSc after scrapie infection where P-tau might be more likely to impact disease course and pathology.
Scrapie infection of transgenic F1 hybrid mice expressing both membrane-anchored and anchorless PrP is known to induce a fatal neurodegenerative disease characterized by deposition of abnormal disease-associated PrPSc as both amyloid and non-amyloid forms.Citation17 In our studies, mice with human tau developed abundant deposits of P-tau in brain by the time of clinical disease. Most P-tau deposits were closely associated with PrPSc amyloid as extracellular aggregates, intraxonal P-tau or perivascular deposits. Intracellular P-tau was also detected with anti-P tau antibody pS396 in scrapie infected mice. Of the 4 anti-tau antibodies tested (CP13, E178, pS396 and pS404), only pS396 stained plaque associated P-tau in huTau− mice which expressed only mouse tau. Perhaps this tau phosphorylation site is more accessible than the others in mouse P-tau using this IHC method. In all areas of co-association of P-tau and PrPSc, there was no obvious increase in neuropathology. In spite of the abundant P-tau deposition observed after scrapie infection, survival curves in F1 mice expressing or not expressing human tau were similar, and if anything mice expressing human tau survived slightly longer (). By analogy, these results suggested that human P-tau might not increase neuropathogenesis in humans with amyloid prion diseases.
There are, however, several other possible explanations for the marginal effects of human tau on survival times in our studies. One option is related to the presence of both amyloid and non-amyloid PrPSc in our mice. Previously the presence of these 2 PrPSc forms had a clear additive effect on the tempo of the clinical disease.Citation17 However, when expressed separately, after scrapie infection these PrPSc forms are associated with distinct diseases differing in neuropathology, tempo and clinical signs.Citation16 The clinical disease associated with the amyloid form of PrPSc is known to be very slow. Therefore, in the current experiments, where the disease tempo was fast, the non-amyloid PrPSc might be the main pathogenic moiety. This interpretation is supported by the finding that the clinical signs observed were nearly identical to those seen in infected mice expressing only wild-type anchored PrP. Possibly human P-tau does not interact sufficiently with non-amyloid PrPSc to alter the disease course. In this case, the interactions which we observed between P-tau and amyloid PrPSc may not have a noticeable effect on the clinical disease because the amyloid-mediated disease is so slow and may not directly contribute to the clinical disease in the dual PrP model. Testing of mice expressing only the anchorless PrP plus human tau might be required to reveal a pathogenic effect of human P-tau in scrapie-induced amyloid prion disease.
Other explanations for the lack of an effect on the survival time in our experiments might be related to P-tau itself. In our model, mice expressed human tau at a level approximately 3-fold above normal, so insufficient tau is unlikely to be an issue. However, the types of P-tau generated might be less pathogenic than expected. Lower pathogenicity might be a result of reduced phosphorylation at specific sites important in eliciting a damage response in brain. Although many reports present evidence for P-tau, it remains unclear which forms are the most pathogenic. The effects of tau oligomer size and fibril size on induction of brain damage is also a topic of current study, and might be influencing our experiments as well. Another issue related to tau itself is the fact that we used a non-mutant form of human tau. Other experiments with an AD model have shown that P301L mutant human tau expressed at higher levels gave a dramatic increase in pathogenesis in APP23 and APP-V717I transgenic mice.Citation21,23 Lastly, we cannot exclude interactions between mouse tau and human tau which might have altered phosphorylation or toxicity of human tau.Citation15,24 More experiments in the future will be required to distinguish among these possibilities.
METHODS
Mice
All mice were housed at the Rocky Mountain Laboratory (RML) in an AAALAC-accredited facility in compliance with guidelines provided by the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research Council). Experimentation followed RML Animal Care and Use Committee approved protocol #2013–064. To create the experimental mice we crossed tg44 mice with htau mice. Tg44 mice (genotype: GPI-anchorless PrP+/+, mouse Prnp−/−, mouse tau+/+, human tau −/−) have a transgene that expresses GPI-anchorless mouse PrP. Tg44 mice express wild-type mouse tau but do not express wild-type mouse PrP or human tau.Citation16 B6.Cg-Mapttm1(EGFP)Klt Tg(MAPT)8cPdav/J (htau) miceCitation15 were obtained from Jackson Laboratories (stock #005491). The htau mice (genotype: GPI-anchorless PrP−/−, mouse Prnp+/+, mouse tau−/−, human tau+/−) do not express GPI-anchorless mouse PrP or mouse tau but they do express wild-type mouse PrP and are hemizygous for a transgene that produces all 6 isoforms of human tau. We mated tg44 mice to htau mice to create F1 offspring. All F1 mice were hemizygous (+/−) for GPI-anchorless PrP, mouse Prnp, and mouse tau. Since the human tau transgene was hemizygous (+/−) in the parental htau mice used, we expected 50 percent of the F1 offspring would carry the human tau transgene and 50 percent would be null for the human tau transgene. All F1 mice were genotyped to determine the carrier status of the human tau transgeneCitation15 and, as expected, half of the pups were hemizygous for the human tau transgene (huTau+) and half were null for the human tau transgene (huTau−). Gender matched littermates representing either huTau+ or huTau− were selected for experimentation.
Inoculations and Clinical Observations
Young adult (6–8 week old) huTau+ and huTau− mice were infected with either RML or 22L rodent-adapted scrapie.Citation16 Each mouse was anesthetized with isoflurane and then injected in the left brain hemisphere with 30 microliters of a 1% scrapie brain homogenate stock diluted in phosphate buffered balanced saline solution + 2% fetal bovine serum. Each 30 microliter inoculum contained either 2.4 × 104 LD50 for RML scrapie or 6.0 × 105 LD50 for 22L scrapie. Following inoculation, mice were monitored for onset of prion disease signs by observers blind to the experimental groups. Mice were euthanized when they displayed advanced stages of prion disease including poor grooming, kyphosis, ataxia, wasting, delayed response to stimuli, and somnolence. Following euthanasia brains were removed, and half of the brain was placed into formalin and half of the brain was flash frozen for biochemical analysis. Survival curve analysis was done using GraphPad PRISM software and the Log-rank (Mantel-Cox) test.
Immunoblotting
For each experimental group, 4-6 mice were analyzed and representative mice are shown in . Brain tissue was homogenized in cell lysis buffer (Bio-Rad) with protease and phosphatase inhibitors as a 20% (wt/vol) brain homogenate (BH) using a mini-bead beater for 45 s on the homogenization setting. Aliquots were stored at −20°C. For PrPres western blots samples were treated with proteinase K (PK) at 50 μg/ml. Briefly 20 μl of a 20% BH from each mouse was adjusted to 100 mM Tris-HCl (pH 8.3), 1% Triton X-100, 1% sodium deoxycholate and 50 μg/ml in a total volume of 31 μl. Samples were treated in a water bath for 45 min at 37°C. All PK digestions were stopped by adding 2 μl of 100 mM Pefabloc (Roche Diagnostics), and the reaction mixture was placed on ice for 5 min. An equal volume of 2 × Laemmli sample buffer (Bio-Rad, Hercules, CA) was added, and then tubes were boiled for 5 min. Samples were frozen at −20°C until they were thawed and re-boiled for 5 minutes, and 4 µl per lane was electrophoresed on a 16% Tris-glycine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel (Life Technologies, CA) and blotted to polyvinylidene difluoride (PVDF) membranes using a 7-min transfer on an iBlot (Life Technologies) device. Immunoblots were probed with the anti-PrP antibody D13 at 1:100.Citation25,26 The secondary antibody was peroxidase-conjugated sheep anti-human IgG (Sigma) at a 1:5,000 dilution. Protein bands were visualized using an enhanced chemiluminescence (ECL) detection system (GE Healthcare) and exposure to film.
For tau immunoblots, 18 µl of 20% BH was brought up to 3.2% SDS and 1× Laemmli buffer (Bio-Rad, Hercules, CA) in a final volume of 90 µl. Each sample was boiled for 5 min and 15 µl per lane was electrophoresed on a 10% Tris-glycine SDS-PAGE gel and blotted on to PVDF membranes using a 7 min transfer on an iBlot. Blots were probed with several anti-tau antibodies. Mouse monoclonal antibody CP27, specific for total human tau, was a gift from Peter Davies and was used at 1:50 dilution. P-tau specific antibodies were as follows: CP13 (Davies), mouse monoclonal antibody specific for pS202 was used at a 1:50 dilution, while rabbit polyclonal antibodies pS396 or pS404 (Invitrogen) were used at 1:1000 and 1:300 respectively. Secondary antibody used for CP27 and CP13 was peroxidase-conjugated rabbit anti-mouse IgG at a 1:5,000 dilution, and secondary antibody for pS396 and pS404 was peroxidase-conjugated goat anti-rabbit IgG used at a 1:3,000 dilution.
Immunohistochemistry
For each experimental group 4-6 half-brains were removed and placed in 10% neutral buffered formalin for 3 to 5 d. Tissues were then processed by dehydration and embedding in paraffin. Sections were cut using a standard Leica microtome, placed on positively charged glass slides, and air-dried overnight at room temperature. The following day slides were heated in an oven at 60°C for 20 min. De-paraffinization, antigen retrieval and staining were performed using the Discovery XT Staining Module and a double stain protocol. Antigen retrieval was achieved using extended cell conditioning with CC1 buffer (Ventana) containing Tris-Borate-EDTA, pH 8.0 for 100 minutes at 95°C as previously described.Citation27 Mouse monoclonal antibody CP13 (anti-pS202), the 1st primary antibody applied was at a dilution of 1:100 in antibody dilution buffer (Ventana) and was incubated for 2 hours at 37°C. The secondary antibody was Biogenex ready-to-use Super Sensitive (SS) Mouse Link (biotinylated goat anti-mouse) (Biogenex, Fremont, CA) used undiluted for 24 min at 37°C. Staining was developed using the DAB Map streptavidin-biotin peroxidase detection system (Ventana). The second primary antibody, D13, a monoclonal anti-PrP antibody, was then applied at a dilution of 1:400 in antibody dilution buffer (Ventana) for 2 hours at 37°C. The secondary antibody, biotinylated goat anti-human IgG (Jackson ImmunoResearch, West Grove, PA) was diluted 1:250 in Ventana antibody dilution buffer and applied for 32 min at 37°C. The second staining was developed using the Red Map fast red detection system (Ventana) and hematoxylin was then used as a counterstain.
For single IHC staining with primary antibodies pS404, pS396 and CP13 an extended cell conditioning step was also performed. Rabbit polyclonal antibody pS404 was used at a 1:100 dilution for 1 hour at 37°C, rabbit polyclonal antibody pS396 was diluted 1:4500 and applied for 1 hour at 37°C. The secondary antibody for both pS404 and pS396 was Biogenex ready-to-use SS Rabbit Link (biotinylated goat anti-rabbit) undiluted and applied for 32 min at 37°C. CP13 single staining followed the same protocol as described above for CP13 for dual staining.
Single IHC staining with total tau antibodies E178 and CP27 used a standard online CC1 antigen retrieval step. Following the cell conditioning slides were stained with rabbit monoclonal antibody E178 a 1:8000 dilution for 2 hours at 37°C. The secondary antibody was the Biogenex ready-to-use SS Rabbit Link undiluted for 20 min at 37°C. Mouse monoclonal antibody CP27 was diluted 1:200 and applied for 2 hours at 37°C followed by the secondary antibody, Biogenex ready-to-use Super Sensitive (SS) Mouse Link for 20 minutes at 37°C. Staining was developed using the DAB Map streptavidin-biotin peroxidase detection system (Ventana).
ABBREVIATIONS
| P-tau | = | phosphorylated tau |
| A-β | = | amyloid β |
| CJD | = | Creutzfeldt-Jakob disease |
| vCJD | = | variant CJD |
| PrP | = | prion protein |
| PrPSc | = | prion disease associated prion protein |
| NFT | = | neurofibrillary tangle |
| AD | = | Alzheimer disease |
| GPI | = | glycosylphosphatidylinositol |
| huTau+ | = | transgenic mice that express human tau |
| huTau− | = | transgenic mice that do not express human tau |
| RML | = | Rocky Mountain Laboratories |
| APP | = | amyloid precursor protein |
| IHC | = | immunohistochemistry |
| LD50 | = | the infectious dose causing disease in 50 percent of inoculated animals |
| BH | = | brain homogenate |
| SDS | = | sodium dodecyl sulfate |
| PK | = | proteinase K |
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
ACKNOWLEDGMENTS
We thank Jim Striebel, Karin Peterson, Jay Carroll and Byron Caughey for critical review of the manuscript; Nancy Kurtz and Lori Lubke for assistance with histology preparation; Jeff Severson for animal husbandry and Peter Davies for the anti-tau antibodies CP13 and CP27.
Funding
This research was supported by the Intramural Research Program of the NIH, NIAID.
REFERENCES
- Kruger L, Mandelkow EM. Tau neurotoxicity and rescue in animal models of human Tauopathies. Curr Opin Neurobiol 2016; 36:52-8; PMID:26431808; http://dx.doi.org/10.1016/j.conb.2015.09.004
- Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 2016; 12:15-27; PMID:26635213; http://dx.doi.org/10.1038/nrneurol.2015.225
- Ittner LM, Gotz J. Amyloid-beta and tau–a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci 2011; 12:65-72; PMID:21193853; http://dx.doi.org/10.1038/nrn2967
- Ittner A, Ke YD, van Eersel J, Gladbach A, Gotz J, Ittner LM. Brief update on different roles of tau in neurodegeneration. IUBMB Life 2011; 63:495-502; PMID:21698753; http://dx.doi.org/10.1002/iub.467
- Reiniger L, Lukic A, Linehan J, Rudge P, Collinge J, Mead S, Brandner S. Tau, prions and Abeta: the triad of neurodegeneration. Acta Neuropathol 2011; 121:5-20; PMID:20473510; http://dx.doi.org/10.1007/s00401-010-0691-0
- Ghetti B, Dlouhy SR, Giaccone G, Bugiani O, Frangione B, Farlow MR, Tagliavini F. Gerstmann-Straussler-Scheinker disease and the Indiana kindred. Brain Pathol 1995; 5:61-75; PMID:7767492; http://dx.doi.org/10.1111/j.1750-3639.1995.tb00578.x
- Giaccone G, Mangieri M, Capobianco R, Limido L, Hauw JJ, Haik S, Fociani P, Bugiani O, Tagliavini F. Tauopathy in human and experimental variant Creutzfeldt-Jakob disease. Neurobiol Aging 2008; 29:1864-73; PMID:17560687; http://dx.doi.org/10.1016/j.neurobiolaging.2007.04.026
- Bons N, Mestre-Frances N, Belli P, Cathala F, Gajdusek DC, Brown P. Natural and experimental oral infection of nonhuman primates by bovine spongiform encephalopathy agents. Proc Natl Acad Sci U S A 1999; 96:4046-51; PMID:10097160; http://dx.doi.org/10.1073/pnas.96.7.4046
- Piccardo P, Cervenak J, Yakovleva O, Gregori L, Pomeroy K, Cook A, Muhammad FS, Seuberlich T, Cervenakova L, Asher DM. Squirrel monkeys (Saimiri sciureus) infected with the agent of bovine spongiform encephalopathy develop tau pathology. J Comp Pathol 2012; 147:84-93; PMID:22018806; http://dx.doi.org/10.1016/j.jcpa.2011.09.004
- Sikorska B, Liberski PP, Sobow T, Budka H, Ironside JW. Ultrastructural study of florid plaques in variant Creutzfeldt-Jakob disease: a comparison with amyloid plaques in kuru, sporadic Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker disease. Neuropathol Appl Neurobiol 2009; 35:46-59; PMID:18513219; http://dx.doi.org/10.1111/j.1365-2990.2008.00959.x
- Lawson VA, Klemm HM, Welton JM, Masters CL, Crouch P, Cappai R, Ciccotosto GD. Gene knockout of tau expression does not contribute to the pathogenesis of prion disease. J Neuropathol Exp Neurol 2011; 70:1036-45; PMID:22002429; http://dx.doi.org/10.1097/NEN.0b013e318235b471
- Chohan MO, Haque N, Alonso A, El-Akkad E, Grundke-Iqbal I, Grover A, Iqbal K. Hyperphosphorylation-induced self assembly of murine tau: a comparison with human tau. J Neural Transm (Vienna) 2005; 112:1035-47.
- Dodart JC, Mathis C, Bales KR, Paul SM. Does my mouse have Alzheimer's disease? Genes Brain Behav 2002; 1:142-55; PMID:12884970; http://dx.doi.org/10.1034/j.1601-183X.2002.10302.x
- Kunzi V, Glatzel M, Nakano MY, Greber UF, Van Leuven F, Aguzzi A. Unhampered prion neuroinvasion despite impaired fast axonal transport in transgenic mice overexpressing four-repeat tau. J Neurosci 2002; 22:7471-7; PMID:12196569
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 2003; 86:582-90; PMID:12859672; http://dx.doi.org/10.1046/j.1471-4159.2003.01879.x
- Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog 2010; 6:e1000800; PMID:20221436
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435-9; PMID:15933194; http://dx.doi.org/10.1126/science.1110837
- Asuni AA, Perry VH, O'Connor V. Change in tau phosphorylation associated with neurodegeneration in the ME7 model of prion disease. Biochem Soc Trans 2010; 38:545-51; PMID:20298219; http://dx.doi.org/10.1042/BST0380545
- Wang GR, Shi S, Gao C, Zhang BY, Tian C, Dong CF, Zhou RM, Li XL, Chen C, Han J, et al. Changes of tau profiles in brains of the hamsters infected with scrapie strains 263 K or 139 A possibly associated with the alteration of phosphate kinases. BMC Infect Dis 2010; 10:86; PMID:20356412; http://dx.doi.org/10.1186/1471-2334-10-86
- Rubenstein R, Chang B, Petersen R, Chiu A, Davies P. T-tau and P-tau in brain and blood from natural and experimental prion diseases. PLoS One 2015; 10:e0143103; PMID:26630676
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 2010; 142:387-97; PMID:20655099; http://dx.doi.org/10.1016/j.cell.2010.06.036
- Denk F, Wade-Martins R. Knock-out and transgenic mouse models of tauopathies. Neurobiol Aging 2009; 30:1-13; PMID:17590238; http://dx.doi.org/10.1016/j.neurobiolaging.2007.05.010
- Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, Van Leuven F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol 2008; 172:786-98; PMID:18258852; http://dx.doi.org/10.2353/ajpath.2008.070904
- Ando K, Leroy K, Heraud C, Kabova A, Yilmaz Z, Authelet M, Suain V, De Decker R, Brion JP. Deletion of murine tau gene increases tau aggregation in a human mutant tau transgenic mouse model. Biochem Soc Trans 2010; 38:1001-5; PMID:20658993; http://dx.doi.org/10.1042/BST0381001
- Rangel A, Race B, Klingeborn M, Striebel J, Chesebro B. Unusual cerebral vascular prion protein amyloid distribution in scrapie-infected transgenic mice expressing anchorless prion protein. Acta Neuropathol Commun 2013; 1:25; PMID:24252347; http://dx.doi.org/10.1186/2051-5960-1-25
- Matsunaga Y, Peretz D, Williamson A, Burton D, Mehlhorn I, Groth D, Cohen FE, Prusiner SB, Baldwin MA. Cryptic epitopes in N-terminally truncated prion protein are exposed in the full-length molecule: dependence of conformation on pH. Proteins 2001; 44:110-8; PMID:11391773; http://dx.doi.org/10.1002/prot.1077
- Klingeborn M, Race B, Meade-White KD, Rosenke R, Striebel JF, Chesebro B. Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol 2011; 85:1484-94; PMID:21123371; http://dx.doi.org/10.1128/JVI.02167-10