
ABSTRACT
Synucleinopathies are a group of neurodegenerative diseases characterized by the accumulation of α-synuclein amyloids in several regions of the brain. α-Synuclein fibrils are able to spread via cell-to-cell transfer, and once inside the cells, they can template the misfolding and aggregation of the endogenous α-synuclein. Multiple mechanisms have been shown to participate in the process of propagation: endocytosis, tunneling nanotubes and macropinocytosis. Recently, we published a research showing that the cellular form of the prion protein (PrPC) acts as a receptor for α-synuclein amyloid fibrils, facilitating their internalization through and endocytic pathway. This interaction occurs by a direct interaction between the fibrils and the N-terminal domain of PrPC. In cell lines expressing the pathological form of PrP (PrPSc), the binding between PrPC and α-synuclein fibrils prevents the formation and accumulation of PrPSc, since PrPC is no longer available as a substrate for the pathological conversion templated by PrPSc. On the contrary, PrPSc deposits are cleared over passages, probably due to the increased processing of PrPC into the neuroprotective fragments N1 and C1. Starting from these data, in this work we present new insights into the role of PrPC in the internalization of protein amyloids and the possible therapeutic applications of these findings.
Introduction
Neurodegenerative disorders are a group of diseases characterized by loss of neuronal cells, synaptic dysfunction and cognitive impairment [Citation1]. These symptoms are associated with the abnormal accumulation of misfolded proteins in the brain and also in peripheral tissues. In synucleinopathies, such as Parkinson's disease (PD), dementia with Lewy bodies (LBD) and multiple system atrophy (MSA), insoluble amyloids of misfolded α-synuclein protein are found throughout the brain, both in neurons (PD and LBD) and glial cells (MSA) [Citation2–4]. It has been shown that, as in the case of prion diseases, also misfolded α-synuclein is able to template the conversion of the endogenous protein in healthy cells and propagate to anatomically connected regions via cell-to-cell transfer [Citation5–8]. Moreover, recent studies pointed out that α-synuclein oligomeric species, rather than large amyloids, are the most toxic species and can be released from neurons, contributing to the spreading of the disease and cognitive decline [Citation9].
Although the precise mechanism by which misfolded α-synuclein spreads and starts the neurodegenerative process remains to be elucidated, great progresses have been done in the last few years that led to the identification of novel pathways and molecular partners involved in the uptake and propagation of the amyloids (see [Citation10] for review). As shown for other proteins (prion protein [Citation11] and tau [Citation12], α-synuclein amyloid fibrils can move from one cell to the other through tunneling nanotubes, membrane structures that connect the cytoplasm of two adjacent cells [Citation13]. Clathrin-dependent endocytosis mediates in part the internalization of the fibrils, and the whole process seems to begin with the interaction between extracellular amyloids and the transmembrane protein lymphocyte-activation gene 3 (LAG3) [Citation14]. This binding is specific to fibrillar forms of α-synuclein, since both monomeric α-synuclein and other fibrillary proteins (tau fibrils, amyloid-ß) do not interact, and the deletion of LAG3 results in a substantial attenuation of cell-to-cell transmission and associated neurotoxicity. In addition to the prominent role of LAG3, synuclein fibrils were shown to interact with other proteins on the surface of the cell, such as neurexin 1 and amyloid β precursor-like protein 1 (APLP1), and with heparin sulfate proteoglycans (HSPGs) that mediate macropinocytosis [Citation15]. Their role as synuclein fibrils-binding partners and their ability to promote the endocytosis of the amyloids is still under evaluation.
It is likely that more than one membrane protein is involved in the internalization of α-synuclein fibrils, and this could help the development of therapeutic approaches by providing a wider spectrum of possible molecules to target in order to reduce the uptake. This is the case of the cellular form of the prion protein (PrPC), which we have recently shown to play a role in the uptake of α-synuclein amyloids [Citation16].
The cellular prion protein acts as a receptor of aggregated proteins
The PrPC is a surface protein anchored to the cell membrane through a C-terminal glycosylphosphatidylinositol (GPI) moiety. Two domains can be clearly identified: the N-terminal, unstructured and positively charged, and the C-terminal, globular and structured (3 α-helices, 2 ß sheets and a short tail) [Citation17]. PrPC plays an important role in several cellular functions, spanning from cell cycle and proliferation to copper homeostasis and neuroprotection [Citation18,Citation19]. Recently, other functions were attributed to PrPC, that is acting as a receptor for amyloid-ß (Aß) oligomers [Citation20] and mediating the Aß- induced synaptic dysfunction. Indeed, it was proposed that, although non-mandatory for the internalization and spreading of the amyloids [Citation20, the expression of PrPC and its interaction with the pathological oligomers is involved in memory impairment, inhibition of long-term potentiation and neuronal death [Citation3]. These findings have a direct impact on the therapeutic approaches to Alzheimer's disease, as it is implied that anti-PrPC antibodies can be used to target PrPC and thus reduce the uptake of amyloid oligomers and the associated neurodegeneration [Citation21].
Given these results, the question arises whether the role of PrPC as a receptor for aggregated proteins can be extended also to other proteins related to neurodegeneration, like α-synuclein and tau. In our recent work, we elucidated some aspects of the complex relationship between α-synuclein and PrPC 16.
In this work was observed that there is a more consistent uptake of fibrillar forms of α-synuclein in the immortalized murine neuroblastoma cell line (N2a) constitutively expressing PrPC (PrP+/+) compared to the same cell line ablated for PrPC (PrP−/−), and also in mouse primary cortical neurons (PrP+/+ and PrP−/−). The sole presence of PrPC is responsible for this difference, since the uptake becomes comparable once its expression is restored. As for Aß-amyloids, also for α-synuclein fibrils the interaction is mediated by a direct binding of the amyloids to the N-terminal part of PrPC, which is known to possess a high-affinity binding site for amyloid structures [Citation22]. The analysis of the dissociation constants (KD) between the two partners calculated by surface plasmon resonance gave two different results (one being around 3 nM, and the other with a binding affinity ten times lower), which can be explained by the fact that the population of fibrils is not homogeneous. Indeed, before using the amyloids in our experiments, α-synuclein fibrils were subjected to sonication, a process that breaks the amyloids randomly to form smaller, oligomeric-like species that are more likely to enter the cells [Citation9]. As such, we hypothesize that smaller fibrillar species establish stronger interactions with PrPC, while long fibrils form weaker interactions, or, alternatively, the binding occurs forming first weak interactions and then stronger interactions, thus explaining the two different values of kD. These findings are in agreement with other studies showing an analogue interaction both in cell lines overexpressing PrPC and in brain slices [Citation23,Citation24]. While previous works pointed at PrPC as the initiator of a signalling cascade that leads to synaptic dysfunction in response to oligomers [Citation24,Citation25], we propose a new model in which the prion protein is directly involved in facilitating the internalization of α-synuclein fibrils; this occurs probably through endocytosis, since in PrPC-expressing cells, fibrils were found predominantly in lysosomes, where all the endocytic pathways converge. In line with its role in the uptake, PrPC promotes also the spreading of the pathology in different brain areas in mice; starting from the injection site in the Substantia Nigra pars compacta (SNpc), after 5 months α-synuclein amyloids were found in the cortex, striatum, thalamus and hippocampus of PrP+/+ mice, together with other key markers of LB pathology. In comparison, PrP−/− mice accumulated less amyloid, both in terms of amount of amyloid deposits and diffusion.
α-Synuclein fibrils interfere with the pathological form of PrP
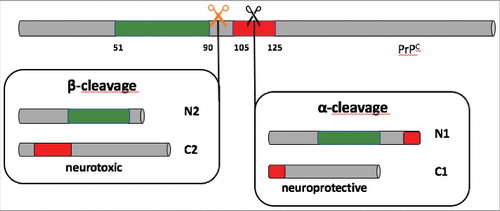
The central event in the pathology of prion diseases is the conversion of PrPC into the insoluble, proteinase-K resistant PrPSc, whose role as the only pathogenic agent has now been widely accepted [Citation26]. However, some cases of prion diseases have been reported in which prions coexist with α-synuclein amyloids, and they are characterized by a longer disease course [Citation27]. This implies that the presence of aggregated α-synuclein in the form of Lewy bodies (LB) and/or Lewy neurites (LN) retard the accumulation and the spreading of PrPSc. One possible explanation relies on the fact that the presence of PrPC molecules on the surface of the cells is essential for the replication of PrPSc, as it acts as a substrate for the formation of new prions. If α-synuclein fibrils are concomitantly present, they compete with PrPSc for the available molecules of PrPC, reducing the pool of substrate and therefore blocking, or at least slowing down, PrPSc replication. At the same time, the interaction with PrPC promotes the propagation of the α-synuclein pathology through the brain. Moreover, α-synuclein amyloids seem to play an additional, active role in prion diseases. Indeed, cell lines permanently infected with prions showed a significant reduction of the amount of PrPSc when treated alongside with several preparations of α-synuclein fibrils. This decrease was accompanied by the appearance of bioactive and neuroprotective fragments N1 and C1 that originate from the α-cleavage of PrPC by members of the protease ADAM family. Cleaving in α-position (between amino acids 110/111 or 111/112) disrupts the site responsible for PrPSc replication and toxicity, and the C1 fragment that is produced has been proved to interfere with the conversion and accumulation of PrPSc 28 ().
Figure 1. Schematic representation of α- and ß- cleavage of the prion protein (PrPC). In green: octapeptide-repeated region; in red: neurotoxic domain; orange and black scissors: cleavage site for ß- and α- cleavage, respectively. ß-cleavage generates fragments N2 and C2, which is neurotoxic; α- cleavage forms the two fragments N1 and C1. α- cleavage destroys the domain responsible for neurotoxicity, and C1 fragment shows neuroprotective properties.
It is noteworthy that among all the amyloids tested, those which were able to lower the PrPSc (α-synuclein, tau K18 fragment) were also associated with the production of the C1 fragment of PrP, while treatment with Aß lead to neither PrPSc decrease nor the production of neuroprotective fragments. This last observation is quite interesting and suggests that the interplay between amyloidogenic proteins in neurodegenerative diseases is very complex and cannot be oversimplified, but needs to be addressed in specific ways, depending on the partners involved. As for α-synuclein, also tau pathology has been shown to occur concomitantly with prion deposits, like in some familial cases of Gerstmann-Sträussler-Scheinker disease (GSS) and in cerebral amyloid angiopathy [Citation29,Citation30]. The relationship between tau and PrPSc has not been clarified yet, but the proposed role of tau fibrils in clearing the PrPSc accumulation opens new interesting perspectives and requires further study.
Conclusions
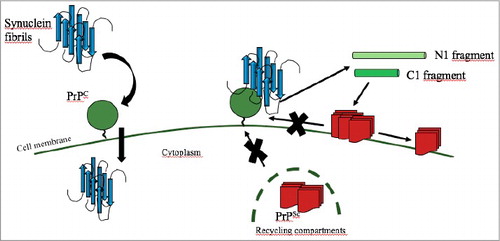
One of the key events in the pathogenesis of neurodegenerative diseases with deposition of misfolded proteins is the ability of the amyloids to enter healthy cells and promote the conversion of the endogenous protein into the aggregation-prone form. In Parkinson's disease, α-synuclein fibrils deposit inside neurons and can be released and taken up by adjacent cells, spreading the pathology and causing neurodegeneration. Several pathways for the uptake of α-synuclein fibrils have been proposed, such as clathrin- and dynamin-mediated endocytosis, micropinocytosis and tunneling nanotubes. However, the extents of which these mechanisms contribute to the internalization, and the involvement of any membrane protein acting as a receptor have not been fully elucidated. In our study [Citation16], we showed that PrPC on the cell surface promotes the uptake of different fibrillar forms of α-synuclein through a direct binding by its N-terminal domain. This interaction is also in part responsible for the reduction of PrPSc in infected cell lines, since PrPC is no longer available for the pathological conversion templated by PrPSc (). The observation that PrPC plays a role in α-synuclein fibrils internalization may have therapeutical applications. Indeed, antibodies against PrPC could be used to target the endogenous PrPC molecules on the cell membrane, thus hampering the interaction with α-synuclein amyloids and reducing cell entry and spreading of synucleinopathies.
Figure 2. Schematic cartoon showing a proposed model for the interaction between PrPC (green circle), PrPSc (red square) and α-synuclein fibrils (in blue). On the left, α-synuclein fibrils bind to PrPC and are internalized by cells. On the right, when α-synuclein fibrils are bound to PrPC, the interaction between PrPC and PrPSc is hampered and the conversion cannot take place. The interaction with α-synuclein fibrils increases the processing of PrPC into the fragment N1 (light green) and C1 (bright green). C1 fragment has a neuroprotective effect and promotes the clearance of the accumulated PrPSc.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Kovacs GG. Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci. 2016;17:189. doi:10.3390/ijms17020189
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi:10.1038/35081564.
- Kostylev MA, Kaufman AC, Nygaard HB, et al. Prion-protein-interacting amyloid-beta oligomers of high molecular weight are tightly correlated with memory impairment in multiple alzheimer mouse models. J Biol Chem. 2015;290:17415–38. doi:10.1074/jbc.M115.643577.
- Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends in neurosciences. 2010;33:317–25. doi:10.1016/j.tins.2010.04.003.
- Luk KC, Song C, O'Brien P, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–6. doi:10.1073/pnas.0908005106.
- Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi:10.1016/j.neuron.2011.08.033.
- Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–5. doi:10.1073/pnas.0903691106.
- Le NT, Narkiewicz J, Aulic S, Salzano G, et al. Synthetic prions and other human neurodegenerative proteinopathies. Virus Res. 2015;207:25–37. doi:10.1016/j.virusres.2014.10.020.
- Lee HJ, Bae EJ, Lee SJ. Extracellular alpha–synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10:92–8. doi:10.1038/nrneurol.2013.275.
- Tyson T, Steiner JA, Brundin P. Sorting out release, uptake and processing of alpha-synuclein during prion-like spread of pathology. J Neurochem. 2016;139(Suppl 1):275–89. doi:10.1111/jnc.13449.
- Zhu S, Victoria GS, Marzo L, et al. Prion aggregates transfer through tunneling nanotubes in endocytic vesicles. Prion. 2015;9:125–35. doi:10.1080/19336896.2015.1025189.
- Abounit S, Wu JW, Duff K, et al. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion. 2016;10:344–51. doi:10.1080/19336896.2016.1223003.
- Abounit S, Bousset L, Loria F, et al. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016;35:2120–38. doi:10.15252/embj.201593411.
- Mao X, Ou MT, Karuppagounder SS, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science (New York, NY). 2016;353: doi:10.1126/science.aah3374.
- Holmes BB, DeVos SL, Kfoury N, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–47. doi:10.1073/pnas.1301440110.
- Aulic S, Masperone L, Narkiewicz J, et al. Alpha-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci Rep. 2017;7:10050. doi:10.1038/s41598-017-10236-x.
- Zahn R, Liu A, Luhrs T, et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000;97:145–50. doi:10.1073/pnas.97.1.145.
- del Rio JA, Gavin R. Functions of the cellular prion protein, the end of Moore's law, and Ockham's razor theory. Prion. 2016;10:25–40. doi:10.1080/19336896.2015.1126038.
- Gasperini L, Meneghetti E, Pastore B, et al. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal. 2015;22:772–84. doi:10.1089/ars.2014.6032.
- Lauren J, Gimbel DA, Nygaard HB, et al. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–32. doi:10.1038/nature07761.
- Barry AE, Klyubin I, Mc Donald JM, et al. Alzheimer's disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:7259–63. doi:10.1523/JNEUROSCI.6500-10.2011.
- Kessels HW, Nguyen LN, Nabavi S, et al. The prion protein as a receptor for amyloid-beta. Nature. 2010;466:E3–4. discussion E-5. doi:10.1038/nature09217.
- Urrea L, Segura-Feliu M, Masuda-Suzukake M, et al. Involvement of Cellular Prion Protein in alpha-Synuclein Transport in Neurons. Mol Neurobiol. 2017. doi:10.1007/s12035-017-0451-4.
- Ferreira DG, Temido-Ferreira M, Miranda HV, et al. Alpha-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci. 2017;20:1569–79. doi:10.1038/nn.4648.
- Um JW, Nygaard HB, Heiss JK, et al. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15:1227–35. doi:10.1038/nn.3178.
- Colby DW, Prusiner SB. Prions. Cold Spring Harbor Perspectives in Biology. 2011;3:a006833. doi:10.1101/cshperspect.a006833.
- Haik S, Privat N, Adjou KT, et al. Alpha-synuclein-immunoreactive deposits in human and animal prion diseases. Acta Neuropathol. 2002;103:516–20. doi:10.1007/s00401-001-0499-z.
- Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J Biol Chem. 2011;286:44234–42. doi:10.1074/jbc.M111.286195.
- Ghetti B, Piccardo P, Spillantini MG, et al. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci U S A. 1996;93:744–8. doi:10.1073/pnas.93.2.744.
- Ghetti B, Tagliavini F, Masters CL, et al. Gerstmann-Straussler-Scheinker disease. II. Neurofibrillary tangles and plaques with PrP-amyloid coexist in an affected family. Neurology. 1989;39:1453–61. doi:10.1212/WNL.39.11.1453.