
ABSTRACT
The prion protein (PrP) is composed of two major domains of similar size. The structured C-terminal domain contains three alpha-helical regions and a short two-stranded beta-sheet, while the N-terminal domain is intrinsically disordered. The analysis of PrP mutants with deletions in the C-terminal globular domain provided the first hint that intrinsically disordered domains are inefficiently transported into the endoplasmic reticulum through the Sec61 translocon. Interestingly, C-terminally truncated PrP mutants have been linked to inherited prion disease in humans and are characterized by inefficient ER import and the formation of neurotoxic PrP conformers. In a recent study we found that the Sec61 translocon in eukaryotic cells as well as the SecY translocon in bacteria is inherently deficient in translocating intrinsically disordered proteins. Moreover, our results suggest that translocon-associated components in eukaryotic cells enable the Sec61 complex to transport secretory proteins with extended unstructured domains such as PrP and shadoo.
Biogenesis and maturation of PrPC
In mammalian cells, biogenesis of PrPC is initiated on free ribosomes in the cytosol. Similarly to other proteins destined for the secretory pathway, PrPC is characterized by a N-terminal signal sequence, which is recognized and bound by the signal recognition particle (SRP). SRP interaction mediates targeting of the ribosome-nascent chain complex to the Sec61 translocon and initiates translocation into the ER lumen.
Co- and post-translational modifications of PrPC are initiated with the cleavage of the N-terminal signal peptide (aa 1–22) and the transfer of core glycans to two asparagines in the C-terminal domain [Citation1] (N180 and N196 in murine PrP, N181 and N197 in human and Syrian hamster PrP). Shortly after PrPC is fully translocated into the ER lumen, a glycosylphosphatidylinositol (GPI) anchor is attached to serine 230 [Citation2]. Via its GPI anchor PrPC is finally targeted to the outer leaflet of the plasma membrane. From the cell surface PrPC can be internalized, a process mediated mainly by the unstructured N-terminus.
Prion protein-induced neurodegeneration – the cytoplasmic connection
The first evidence that PrP can acquire a neurotoxic activity due to impaired translocation into the ER lumen emerged from pioneering studies on the biogenesis of wildtype (wt) PrP. These studies revealed that in addition to the fully translocated form PrP can adopt two different transmembrane topologies during ER import, termedNtmPrP (N-terminus facing the ER lumen) or CtmPrP (C-terminus facing the ER lumen) [Citation3]. Under these conditions either the C-terminal or N-terminal part of PrP is present the cytosol. Remarkably, increased synthesis of CtmPrP coincides with progressive neurodegene-ration both in Gerstmann-Sträussler-Scheinker syndrome (GSS) patients and in transgenic mice [Citation4]. The neurotoxic potential of cytosolic PrP conformers (cytoPrP) was further established by expressing a PrP mutant lacking the N-terminal ER targeting signal in transgenic mice and cultured cells [Citation5,Citation6]. Brain extracts from clinically ill transgenic mice expressing cytoPrP do not contain infectious prions, a phenomenon also seen in other transgenic mouse models of neurotoxic PrP mutants (rev. in [Citation7]). Different mechanisms have been described to explain the toxic effects of PrP in the cytosolic compartment. In one study co-aggregation of misfolded cytoPrP with the anti-apoptotic protein Bcl-2 was shown to coincide with toxicity [Citation8]. Another study reported that cytoPrP can interact with the E3 ubiquitin ligase Mahogunin, thereby disrupting its function [Citation9]. Interestingly, a toxic potential was also observed for cytosolically localized PrPSc, which can inhibit proteasomal activity [Citation10]. Cytosolically localized PrP was described to be present in a subset of neurons in mice even under physiological conditions [Citation11].
The structured C-terminal domain of PrP promotes ER import
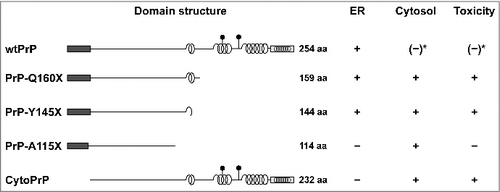
PrP has a particular modular composition (): The N-terminal domain of PrP spanning 120 amino acids is flexibly disordered followed by a highly structured C-terminal domain of approximately 110 amino acids that contains three α-helical regions and a short, two-stranded β-sheet [Citation12–14]. The analysis of PrP-W145X, a pathogenic PrP mutant linked to Gerstmann-Sträussler-Scheinker syndrome in humans, provided the first hint that deletions in the C-terminal domain impair ER import of PrP [Citation15]. We corroborated and extended these studies and showed that impaired ER import is a general phenomenon of C-terminal deletion mutants of PrP linked to inherited prion diseases in humans and that the cytosolically localized PrP species are toxic to cells [Citation6,Citation16]. Furthermore, it appears that the toxic activity resides in the structured domain: Whereas PrP-Y145X and PrP-Q160X induced cell death, a shorter mutant composed of only the unstructured domain (PrP-A115X) did not [Citation6,Citation16] (). Indeed, in a large-scale study three individuals were identified expressing PrP-Q75X or PrP-R37X. They were free of overt neurological disease at ages 79, 73, and 52 and report no personal or family history of neurodegeneration or peripheral neuropathy [Citation17].
Figure 1. Intracellular localization of PrP and neurotoxic properties in mammalian cells. Schematic presentation of PrP and mutants thereof. Dark rectangle: signal peptide; α-helical structure is indicated by helices, intrinsically disorder by a straight line; polygons represent N-linked glycosylation acceptor site; white rectangle: GPI anchor signal sequence. Please note that GPI signal peptides are predicted to adopt an alpha-helical structure. Neurotoxic PrP mutants linked to inherited prion diseases in humans (PrP-Q160X and PrP-Y145X) are characterized by deletions in the C-terminal globular domain and impaired import into the ER. Expression of cytoPrP in transgenic mice induces severe ataxia due to rapid cerebellar granule neuron degeneration. *It was described that also wildtype PrP can remain in the cytosol and interfere with cellular viability. For details see main text.
Employing neuropeptide hormones and model substrates we could then show that impaired ER import of intrinsically disordered proteins (IDPs) is a general phenomenon and not a specific feature of PrP mutants. Specifically, these studies revealed that IDPs require alpha-helical domains in addition to the N-terminal signal peptide for efficient Sec61-mediated transport into the ER [Citation16,Citation18–21].
The Sec61/SecY complex is inherently deficient in translocating intrinsically disordered proteins
The Sec61 complex in eukaryotes is highly homologous to the SecY complex, which mediates translocation of secretory proteins into the periplasm of bacteria. In addition, the co-translational targeting pathway mediated by SRP is conserved (rev. in [Citation22) (]). Thus, we were wondering whether impaired transport of IDPs is a general phenomenon of Sec61/SecY-mediated translocation. To address this question, we performed a comparative analysis and studied transport of IDPs through the Sec61 complex in mammalian and yeast cells and through the SecY complex in E. coli [Citation23]. The model substrates were modified with authentic N-terminal signal peptides of the respective organisms. Mammalian proteins contained the signal peptide of mouse PrP (GenBank accession number M18070) or of rat growth hormone (NCBI Reference Sequence NC_005109.4), for secretion in E. coli, the substrates were equipped with the DsbA signal peptide (NCBI Reference Sequence NP_418297.1) for targeting via the SRP-dependent pathway (mainly co-translational, see above). In addition, the PelB signal peptide (GenBank Reference Sequence M17364.1) was used to target the substrates to a post-translation pathway specific for E. coli (prokaryotes). Under these conditions the proteins are first bound by a chaperone (SecB) after they have been released form free ribosomes in the cytosol and translocation through the SecY complex is then driven by an ATPase (SecA) [Citation24]. For our studies in Saccharomyces cerevisiae the Kre5p signal peptide (NCBI Reference Sequence NM_001183756.1) was used. Productive ER import in eukaryotic cells was monitored by the appearance of a glycosylated protein fraction, since all constructs contained acceptor sites for N-linked glycosylation. In E. coli transport of the constructs into the periplasm was analyzed by separating transformed bacteria into periplasmic and cytoplasmic fractions and subsequent Western blotting. Indeed, our studies revealed an impairment of both the Sec61 translocon in eukaryotes and the SecY translocon in E. coli to translocate proteins that are entirely disordered. This impairment seemed to be independent of the length of the protein, the signal peptide used and the targeting mode (co- or post-translationally) [Citation23] (). The unstructured substrates that failed to be imported contained an uncleaved signal peptide and were subjected to proteasomal degradation in mammalian cells. Thus, we disfavor an interpretation that the proteins were first imported into the ER lumen, which would result in signal peptide cleavage, and then subjected to the ER-associated degradation (ERAD) pathway. We rather assume that targeting of the ribosome-nascent chain complex to the translocon is not affected, but that after the initial gating by the signal peptide additional alpha-helical domains are required to keep the pore open and ensure efficient translocation into the ER lumen.
Figure 2. Conserved pathways for protein translocation in eukaryotes and prokaryotes. Schematic view of Sec61- and SecY-mediated secretion in eukaryotes and gram-negative bacteria. After targeting of the ribosome-nascent chain complex to the translocon the protein is translocated into the ER lumen or periplasm. In addition to the co-translational targeting pathway depicted in the scheme, substrates can also be targeted after they have been synthesized on free ribosomes in the cytosol (post-translational pathway).
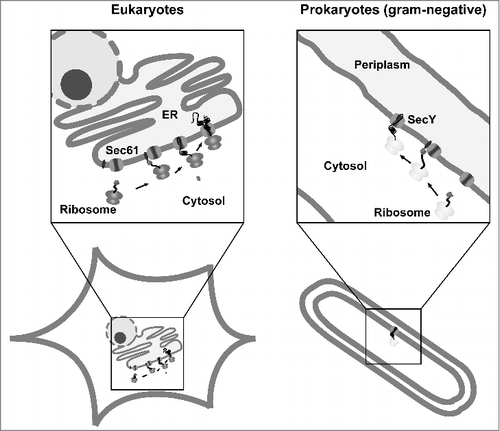
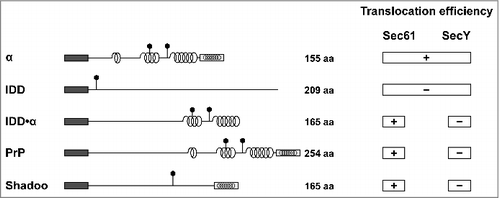
Figure 3. Selective impairment of the SecY complex to translocate secretory proteins with extended intrinsically disordered domains (IDDs) The scheme summarizes the translocation efficiency of different model substrates and natural occurring proteins, such as PrP and shadoo, into the ER of mammalian and yeast cells (Sec61) or the periplasm of E. coli (SecY). Dark rectangle: signal peptide; α-helical structure is indicated by helices, intrinsically disorder by a straight line; polygons represent N-linked glycosylation acceptor site; white rectangle: GPI anchor signal sequence. Please note that GPI signal peptides are predicted to adopt an alpha-helical structure.
Adaptive pathways in eukaryotic cells enable Sec61-mediated translocation of secretory proteins with extended N-terminal unstructured domains
By including substrates in our analysis that are composed of a large intrinsically disordered domain (IDD) followed by an alpha-helical domain (IDD·α) we found an interesting selective impairment of the SecY complex in E. coli. In mammalian and yeast cells ER import of the IDD was restored by alpha-helical domains. In E. coli, however, a C-terminally located alpha-helical domain was not able to promote SecY-mediated translocation of the IDD ().
Intrigued by the impaired translocation of the artificial IDD·α constructs we analyzed secretion of two naturally occurring mammalian proteins in E. coli that are characterized by extended unstructured domains: PrP and shadoo. Shadoo is a highly conserved neuronal glycoprotein with a stress-protective activity present in all vertebrates [Citation21,Citation25,Citation26]. Indeed, neither PrP nor shadoo equipped with either the DsbA or PelB signal peptides were secreted into the periplasm of bacteria (). Notably, the apparent translocation deficiency is probably of little relevance to bacteria, since secretory proteins with extended N-terminal unstructured domains have not been described so far in prokaryotes. However, also a small fraction of full length wildtype PrP was found in the cytosol of mammalian cells after proteasomal inhibition. This fraction might be generated via retro-translocation of PrP from the ER lumen or be due to an abrogated ER import [Citation27,Citation28]. Moreover, impaired ER import of wildtype PrP seems to be aggravated under stress conditions, leading to enhanced formation of cytosolic PrP with neurotoxic properties (rev. in [Citation29]).
An attractive hypothesis to explain the ability of eukaryotic cells to transport client proteins with IDDs is that specific translocon-associated factors are missing in E. coli. These adaptive mechanisms may have evolved in parallel with the expansion of IDDs in the proteome of eukaryotic cells [Citation30] in order to compensate for the inherent translocation deficiency of the Sec61 complex. It will now be interesting to identify these putative translocon-associated factors and to study their role in the regulation of translocation efficiency in general and specifically in the formation of neurotoxic conformers.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Haraguchi T, Fisher S, Olofsson S, et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch Biochem Biophys. 1989;274:1–13. doi:10.1016/0003-9861(89)90409-8
- Stahl N, Borchelt DR, Hsiao K, et al. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–40. doi:10.1016/0092-8674(87)90150-4
- Yost CS, Lopez CD, Prusiner SB, et al. Non-hydrophobic extracytoplasmic determinant of stop transfer in the prion protein. Nature. 1990;343:669–72. doi:10.1038/343669a0
- Hegde RS, Mastrianni JA, Scott MR, et al. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–34. doi:10.1126/science.279.5352.827
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–5. doi:10.1126/science.1073725
- Rambold AS, Miesbauer M, Rapaport D, et al. Association of Bcl-2 with misfolded prion protein is linked to the toxic potential of cytosolic PrP. Mol Biol Cell. 2006;17:3356–68. doi:10.1091/mbc.E06-01-0083
- Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: Gain and loss of function in neurodegenerative diseases. EMBO J. 2008;27:336–49. doi:10.1038/sj.emboj.7601930
- Rambold AS, Miesbauer M, Rapaport D, et al. Association of Bcl-2 with misfolded prion protein is linked to the toxic potential of cytosolic PrP. Mol Biol Cell. 2006;17:3356–68. doi:10.1091/mbc.E06-01-0083
- Chakrabarti O, Hegde RS. Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell. 2009;137:1136–47. doi:10.1016/j.cell.2009.03.042
- Kristiansen M, Deriziotis P, Dimcheff DE, et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell. 2007;26:175–88. doi:10.1016/j.molcel.2007.04.001
- Mironov AJ, Latawiec D, Wille H, et al. Cytosolic prion protein in neurons. J Neurosci. 2003;23:7183–93.
- Donne DG, Viles JH, Groth D, et al. Structure of the recombinant full-length hamster prion protein PrP(29-231): the N terminus is highly flexible. Proc Natl Acad Sci USA. 1997;94:13452–57. doi:10.1073/pnas.94.25.13452
- Riek R, Hornemann S, Wider G, et al. NMR structure of the mouse prion protein domain PrP(121-321). Nature. 1996;382:180–2. doi:10.1038/382180a0.
- Riek R, Hornemann S, Wider G, et al. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett. 1997;413:282–8. doi:10.1016/S0014-5793(97)00920-4
- Zanusso G, Petersen RB, Jin T, et al. Proteasomal degradation and N-terminal protease resistance of the codon 145 mutant prion protein. J Biol Chem. 1999;274:23396–404. doi:10.1074/jbc.274.33.23396
- Heske J, Heller U, Winklhofer KF, et al. The C-terminal domain of the prion protein is necessary and sufficient for import into the endoplasmic reticulum. J Biol Chem. 2004;279:5435–43. doi:10.1074/jbc.M309570200
- Minikel EV, Vallabh SM, Lek M, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8:322.ra9. doi:10.1126/scitranslmed.aad5169
- Miesbauer M, Pfeiffer NV, Rambold AS, et al. Alpha-helical domains promote translocation of intrinsically disordered polypeptides into the endoplasmic reticulum. J Biol Chem. 2009;284:24384–93. doi:10.1074/jbc.M109.023135
- Miesbauer M, Rambold AS, Winklhofer KF, et al. Targeting of the prion protein to the cytosol: mechanisms and consequences. Curr Issues Mol Biol. 2010;12:109–18.
- Dirndorfer D, Seidel RP, Nimrod G, et al. The alpha-helical structure of prodomains promotes translocation of intrinsically disordered neuropeptide hormones into the endoplasmic reticulum. J Biol Chem. 2013;288:13961–73. doi:10.1074/jbc.M112.430264
- Pfeiffer NV, Dirndorfer D, Lang S, et al. Structural features within the nascent chain regulate alternative targeting of secretory proteins to mitochondria. EMBO J. 2013;32:1036–51. doi:10.1038/emboj.2013.46
- Rapoport TA, Li L, Park E. Structural and Mechanistic Insights into Protein Translocation. Annual review of cell and developmental biology. 2017;33:369–90. doi:10.1146/annurev-cellbio-100616-060439
- Gonsberg A, Jung S, Ulbrich S, et al. The Sec61/SecY complex is inherently deficient in translocating intrinsically disordered proteins. J Biol Chem. 2017. doi:10.1074/jbc.M117.788067
- Mori H, Ito K. The Sec protein-translocation pathway. Trends Microbiol. 2001;9:494–500. doi:10.1016/S0966-842X(01)02174-6
- Daude N, Ng V, Watts JC, et al. Wild-type Shadoo proteins convert to amyloid-like forms under native conditions. J Neurochem. 2010;113:92–104. doi:10.1111/j.1471-4159.2010.06575.x
- Watts JC, Drisaldi B, Ng V, et al. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J. 2007;26:4038–50. doi:10.1038/sj.emboj.7601830
- Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci U S A. 2001;98:14955–60. doi:10.1073/pnas.011578098
- Drisaldi B, Stewart RS, Adles C, et al. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem. 2003;278:21732–43. doi:10.1074/jbc.M213247200
- Chakrabarti O, Ashok A, Hegde RS. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem Sci. 2009;34:287–95. doi:10.1016/j.tibs.2009.03.001
- Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30:137–49. doi:10.1080/07391102.2012.675145