
ABSTRACT
Although colocalization of amyloid β (Aβ) with prion protein (PrP) in the kuru plaque has previously been observed in the brain of prion diseases patients, the participating Aβ species has not been identified. Here, we present an immunohistochemical assessment of the brain and spinal cord of a 69-year-old Japanese female patient with Gerstmann-Sträussler-Scheinker disease with a P105L mutation on the PRNP gene (GSS-P105L). Immunohistochemical assessment of serial brain sections was performed using anti-PrP and -Aβ antibodies in the hippocampus, frontal and occipital lobes. She died 69 years after a 21-year clinical course. Immunohistochemistorical examination revealed that ~50% of the kuru plaques in the cerebrum were colocalized with Aβ, and Aβ42 was predominantly observed to be colocalized with PrP-plaques. The Aβ deposition patterns were unique, and distinct from diffuse plaques observed in the normal aging brain or Alzheimer’s disease brain. The spinal cord exhibited degeneration in the lateral corticospinal tract, posterior horn, and fasciculus gracilis. We have demonstrated for the first time that Aβ42, rather than Aβ40, is the main Aβ component associated with PrP-plaques, and also the degeneration of the fasciculus gracilis in the spinal cord in GSS-P105L, which could be associated with specific clinical features of GSS-P105L.
Introduction
Gerstmann-Sträussler-Scheinker disease (GSS) is an inherited human prion disease characterized by progressive dementia, pyramidal signs and cerebellar ataxia [Citation1]. A substitution of proline for leucine at codon 105 (P105L) has been reported only in Japan, and thus far, several central nervous system neuropathological features have been clarified by neuropathological examinations of seven autopsied cases [Citation2–Citation8].
A few researchers have observed colocalization of amyloid β (Aβ) with prion protein (PrP) in the brain of GSS and familial Creutzfeldt-Jakob disease (fCJD) [Citation8–Citation12], but the predominant PrP-associated amyloid species has not yet been identified. Here, we present detailed immunohistochemical findings of PrP and Aβ in the brain in a 69-year-old Japanese female patient with GSS with a P105L mutation (GSS-P105L), and also the neuropathological features in the spinal cord of our patient.
Results
Case report
The unique symptoms of this patient were reported as ‘sensory-psychiatric symptoms’ in 2002 [Citation13]. Her symptoms presented at the age of 48 years as chest and abdominal pain that migrated to various regions of her body. She underwent various tests and radiological examinations to identify the origin of her pain, but the cause was unclarified. At the age of 53 years, she developed cognitive impairment and gait disturbance, and the following year, she developed confusion. Her first neurological examination performed at the age of 55 years, revealed cognitive impairment, hyper-reflexia with bilateral Babinski signs, and extrapyramidal and frontal lobe signs without cerebellar symptoms. At the age of 57years, she could not sit or speak. Two years later, she could not consume food orally, and we started tube feeding. She died of aspiration pneumonia at 69 years of age. This patient exhibited extremely slow clinical progression, and the disease duration from onset to death was 21 years.
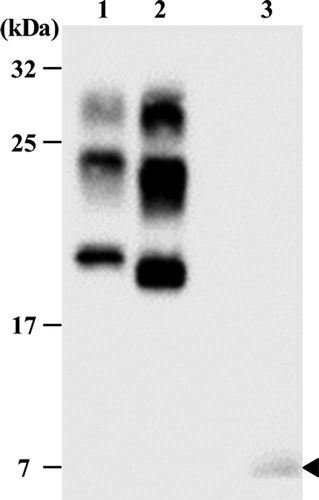
Radiological examinations revealed predominant bilateral atrophy of the frontal lobes and no signal change in diffusion-weighted magnetic resonance imaging (MRI). Electroencephalography indicated diffuse slowing without periodic sharp-wave complexes. The PrP gene analysis revealed a heterozygous missense mutation at codon 105 to leucine and methionine with valine heterozygosity at codon 129 of the same allele. Another polymorphism at codon 219 exhibited glutamic acid homozygosity. The apolipoprotein E genotype was epsilon3/epsilon3. The autopsied brain homogenate was analyzed by western blotting and detected a low molecular weight (MW) band at approximately 7 kDa without a high MW band ().
Figure 1. Western blots analysis of the brain homogenates of the patient, after proteinase K treatment. Brain homogenates from one patient with sporadic Creutzfeldt-Jakob (CJD) with methionine homozygosity type 1 (lane1), one patient with sporadic CJD with methionine homozygosity type 2 (lane2), and our patient (lane3) were treated with proteinase K and underwent sodium dodecyl sulfate-polyacrylamide gel electrophoresis. A low molecular weight band at approximately 7 kDa was detected without a high molecular weight band in our patient (arrow head).
Neuropathological findings
The brain was atrophic, and the weight was 853 g. The cerebral white matter was remarkably atrophic. The pyramis, cerebral peduncle, and cervical to lumbar spinal cord were also atrophic. Microscopically, severe vacuolations, likely caused by ischemia, were observed in the 2nd and 3rd layers of the cerebral cortex, and neuronal loss and gliosis were observed in the deep layers. No spongiform changes were observed in any region of brain. In the cerebral white matter, axonal degenerations accompanied by numerous macrophages were observed. The hippocampus, striatum, amygdala and cerebellum were preserved. The spinal cord exhibited remarkable neuronal degeneration in the lateral corticospinal tract, fasciculus gracils and posterior horn ()).
Figure 2. Neuropathological data in this patient. (a) Degeneration of the posterior horn (arrow head) and fasciculus gracilis (arrow) were observed in spinal cord. (b) Synaptic prion protein (PrP) depositions are detected in posterior horn. Immunostaining with antibodies against PrP (c, f, i), Aβ42 (d, g, j), Aβ40 (e, h, k) was performed in the same location in serial brain sections. We detected three types of PrP-amyloyd β (Aβ) colocalized plaques; (c–e) overlapped type, (f–h) surrounded type, and (i–k) a unique deposition type, wherein Aβ42 deposits were sandwiched by PrP deposits. Aβ42 deposition was observed in all types (d, g, j), but Aβ40 was barely detected (e, h, k). Scale bar = 20μm.
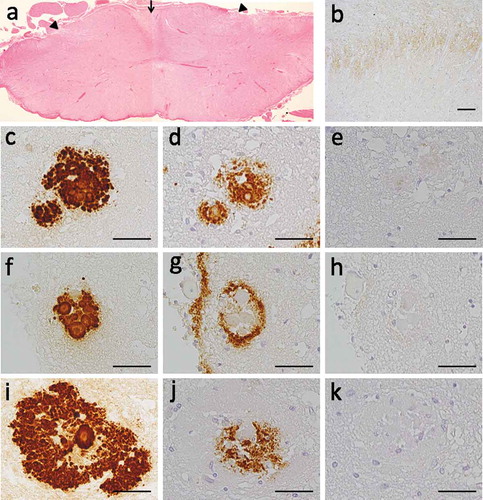
Immunohistochemical examination using the 3F4 antibody revealed a diffuse multicentric core and granular plaques primarily at the cortices, and unicentric core plaques primarily at the borderline between the white matter and gray matter of the cortices. Similar findings were observed in the hippocampus, striatum and amygdala. The posterior horn of the spinal cord exhibited synaptic deposition of PrPs ()).
Immunostaining for both PrP and Aβ using serial brain sections revealed PrP plaques colocalized with Aβ (). The primary Aβ species component associated with PrP-plaques was Aβ42, and deposits of Aβ40 were barely observed (). Three patterns of colocalization of both PrP and Aβ deposits were observed. First, the distribution of Aβ42 deposits overlapped with those of PrP deposits (overlap type, ). Second, diffuse Aβ42 deposits were located around the PrP core (surrounded type, ). Third, Aβ42 deposits were located around PrP core, and these Aβ42 deposits were sandwiched by further PrP deposits (). The amounts of PrP-Aβ colocalized plaques and non-colocalized plaques were similar. In all amyloid plaque deposition, the overlap type was the most frequent deposition pattern (34%). PrP deposits around Aβ cores or diffuse plaques were not observed. The immunoreactive pattern of plaques with anti-Aβ antibody was distinct from the classical senile plaques observed in Alzheimer’s disease (AD), and was dissimilar to the dense core plaques observed in normal aging.
Discussion
To the best of our knowledge, there are no previous reports on GSS-P105L that have identified the PrP plaque-associated Aβ species in GSS and fCJD. Our data clearly demonstrated that the main component of Aβ-plaques associated with PrP-plaques was Aβ42, rather than Aβ40. We report another significant novel finding associated with GSS-P105L, which was degeneration of the fasciculus gracilis in the spinal cord.
Colocalization of PrP and Aβ in the same kuru plaques in patients with GSS was previously described [Citation9], including various type of GSS, such as GSS-P105L [Citation8], GSS-A117V [Citation10] or GSS-P102L [Citation11], and fCJD with E200K mutations [Citation12]. Aβ deposits were primarily observed to be surrounding PrP deposits, but PrP deposits were never located around the Aβ deposits. Our findings indicate a novel colocalized-plaque pattern wherein Aβ42 deposits were located around a PrP core, and were further surrounded by another PrP deposit (,j)), and the frequency of this deposition pattern was very low, accounting for only 0.6% of all amyloid plaques.
Distinct from the diffuse plaque observed in the normal aging brain, the Aβ42-plaques, which overlapped and surrounded PrP-plaques, were predominantly detected. The ‘surrounded type’ deposition pattern of Aβ42 was apparently different from that of diffuse plaques.
Because the ratio of Aβ42:Aβ 40 in the brain was reported to be approximately 1:10 in non-AD, the typical ratio of Aβ42:Aβ40 in the plaques of this patient indicates unnatural processing of Aβ in the brain of GSS patients. We speculate that the possible reason for colocalization is that the preceding PrP deposition may be a type of tropism effect for Aβ42 by PrP deposition. In a recent study, the PrP was reported to possess a binding site for Aβ-oligomers, and the binding site was considered to be the 95–105 segment of cellular PrP [Citation14]. It is possible that the conformational change from normal PrP (P105L) to abnormal PrP (P105L) may strengthen the affinity between PrP and Aβ42.
Aβ42 is known to have high aggregability, and the binding of PrP scrapie (PrPSc) (P105L) may causes a status pathway change of PrP to the less toxic form, which was hypothesized to be an Aβ oligomer in AD pathology [Citation15]. If so, colocalization of PrP and Aβ42 possibly causes a decrease in the very high toxicity of PrPSc. Indeed, the disease duration in our patient was extremely long, which may have been associated with colocalization of PrP and Aβ42 in the brain.
To date, neuropathological analyses of the spinal cords of GSS-P105L patients have been performed [Citation2,Citation5–Citation8], and degeneration of the corticospinal tracts and small PrP depositions in the spinal posterior horn have been reported [Citation2]. The pathological findings in our patient were compatible with such previous findings, and our patient exhibited remarkable neuronal degeneration in the fasciculus gracils throughout the spinal cord, which has not been reported previously, and indicates the possibility of degeneration of the fasciculus gracilis in GSS.
In summary, we firstly determined that Aβ42 was the main component of Aβ associated with PrP-plaques, and also demonstrated of the fasciculus gracilis in patient with GSS. These findings are highly relevant to the central nervous system pathological process in GSS.
Materials and methods
We prepared sections of formalin-fixed, paraffin-embedded brain tissue. Tissue specimens were obtained from the frontal, temporal, and occipital lobes, and the striatum, pallidum, hippocampus, thalamus, cerebellum, brainstem and spinal cord for hematoxylin & eosin staining and immunohistochemical analysis of PrP and Aβ deposits. Immunohistochemical evaluation of the serial hippocampus and frontal and occipital lobe sections involved staining with mouse monoclonal antibody 3F4 (1:500, Signet Laboratories, catalog number 9620-02), 4G8 (1:5000, Covance Inc., catalog number SIG-39220), and anti-Aβ40 (1:1000, Immuno-Biological Laboratories, catalog number 10047), and anti-rabbit Aβ42 antibodies (1:100, Immuno-Biological Laboratories, catalog number 18582). The genetic and western blot analyses were performed as previously described [Citation2,Citation7].
The study protocol followed ethical requirements and was approved by the Institutional Ethics Committee of Tokyo Medical and Dental University. This study was performed in accordance with the ethical standards laid down by the 2013 Declaration of Helsinki.
Author contributions
Fumiko Furukawa participated in the drafting of the manuscript, data collection, and data analysis. Nobuo Sanjo participated in revising the manuscript, the design and conceptualization of the study, data collection, data analysis, and supervised the study. Atsushi Kobayashi performed data collection and analysis, and participated in the conceptualization of the study and revision of the manuscript. Tsuyoshi Hamaguchi performed data analysis. Masahito Yamada participated in the revision of the manuscript and supervised the study. Tetsuyuki Kitamoto performed data collection and analysis, participated in the conceptualization of the study, and supervised the analyses. Hidehiro Mizusawa supervised the study and participated in the revision of the manuscript. Takanori Yokota supervised the study and participated in the revision of the manuscript.
Acknowledgments
The authors thank members of the Department of Neurology and Neurological Science, Tokyo Medical and Dental University hospital, as well as the patient and her family members for providing important clinical information. We are grateful to the member of the Department of Neurology and Neurobiology of Aging, Kanazawa University Graduate School of Medical Science and the Department of Neurological Science, Tohoku University Graduate School of Medicine for their technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Farlow MR, Yee RD, Dlouhy SR, et al. Gerstmann-Sträussler-Scheinker disease. I. Extending the clinical spectrum. Neurology. 1989;39(11):1446–1452.
- Yamada M, Itoh Y, Inaba A, et al. An inherited prion disease with a PrP P105L mutation: clinicopathologic and PrP heterogeneity. Neurology. 1999;53(1):181–188.
- Itoh Y, Yamada M, Hayakawa M, et al. A variant of Gerstmann-Sträussler-Scheinker disease carrying codon 105 mutation with codon 129 polymorphism of the prion protein gene: a clinicopathological study. J Neurol Sci. 1994;127(1):77–86.
- Amano N, Yagishita S, Yokoi S, et al. Gerstmann-Sträussler syndrome? A variant type: amyloid plaques and Alzheimer’s neurofibrillary tangles in cerebral cortex. Acta Neuropathol. 1992;84(1):15–23.
- Isshiki T, Minagawa M, Yamauchi Y. [Spastic paraparesis type of GSS]. Dementia Jpn (Tokyo). 1994;8: 405–411. Japanese.
- Nakazato Y, Ohno R, Negishi T, et al. An autopsy case of Gerstmann-Strӓussler-Scheinker’s disease with spastic paraplegia as its principle feature. Rinsho Shinkeigaku. 1991;31(9):987–992.
- Kitamoto T, Amano N, Terao Y, et al. A new inherited prion disease (PrP-P105L mutation) showing spastic paraparesis. Ann Neurol. 1993;34(6):808–813.
- Yamazaki M, Oyanagi K, Mori O, et al. Variant Gerstmann-Sträussler syndrome with the P105L prion gene mutation: an unusual case with nigral degeneration and widespread neurofibrillary tangles. Acta Neuropathol. 1999;98(5):506–511.
- Miyazono M, Kitamoto T, Iwaki T, et al. Colocalization of prion protein and beta protein in the same amyloid plaques in patients with Gerstmann-Sträussler syndrome. Acta Neuropathol. 1992;83(4):333–339.
- Tranchant C, Sergeant N, Wattez A, et al. Neurofibrillary tangles in Gerstmann-Sträussler-Scheinker syndrome with the A117V prion gene mutation. J Neurol Neurosurg Psychiatry. 1997;63(2):240–246.
- Piccardo P, Ghetti B, Dickson DW, et al. Gerstmann-Sträussler-Scheinker disease (PRNP P102L): amyloid deposits are best recognized by antibodies directed to epitopes in PrP region 90-165. J Neuropathol Exp Neurol. 1995;54(6):790–801.
- Ghoshal N, Cali I, Perrin RJ, et al. Codistribution of amyloid beta plaques and spongiform degeneration in familial Creutzfeldt-Jakob disease with the E200K-129M haplotype. Arch Neurol. 2009;66(10):1240–1246.
- Shiraishi A, Mizusawa H, Yamada M. Early and persistent sensory-psychiatric symptoms in an inherited prion disease with a PrP P105L mutation. J Neurol. 2002;249(12):1740–1741.
- Laurén J, Gimbel DA, Nygaard HB, et al. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457(7233):1128–1132.
- Verma M, Vats A, Taneja V. Toxic species in amyloid disorders: oligomers or mature fibrils. Ann Indian Acad Neurol. 2015;18(2):138–145.