
ABSTRACT
Normal monomeric tau can be converted into pathogenic aggregates and acquire protease resistance in a prion-like manner. This acquisition of partial protease-resistance in tau aggregates has to date only been partially investigated in various studies exploring the prion-like properties of tau. In this study, we induced the aggregation of tau repeat domain (RD) in cultured cells using detergent insoluble fractions of Alzheimer’s brain tissue as seeds. The seeded aggregation of tau RD in cultured cells formed a ~7 kDa protease-resistant fragment in contrast to the ~12 kDa tau fragment characteristic of the AD seeds, suggesting that the in vitro generated tau aggregates were conformationally distinct from parent seeds.
Introduction
Tau is predominantly found in neuronal axons [Citation1] bound to microtubules, helping to stabilize their polymeric structures [Citation2]. There are six isoforms of tau in the adult human brain, each arising from alternative splicing of a single gene, microtubule-associated protein tau (MAPT) on chromosome 17 [Citation3]. The alternative splicing of exons 2 and 3 results in the presence of zero, one, or two insertions (0N, 1N or 2N, respectively) in the amino-terminal region, whereas the alternative splicing of exon 10 results in three repeats (3R) or four repeats (4R) in the carboxyl-terminal repeat domain (RD) [Citation3]. The C-terminal repeat domain of tau binds to microtubules (MT), thereby helping to promote their assembly [Citation4]. The length of tau ranges from 352 to 441 amino acids, depending on the number of N-terminal inserts and on the number of tandem repeats.
Many neurodegenerative diseases are characterized by the deposition of intracellular neurofibrillary tangles and/or neuropil threads, composed of hyperphosphorylated tau assemblies [Citation5]. Those disorders, collectively referred to as tauopathies, include Alzheimer’s disease (AD), frontotemporal dementia with parkinsonism linked to chromosome 17, corticobasal degeneration, and progressive supranuclear palsy [Citation6]. Cognitive deficits in AD closely correlate with the presence of tau pathology in brains [Citation7,Citation8]. Filamentous tau deposits have been shown to propagate along neuroanatomically connected brain regions in a hierarchical manner [Citation5,Citation9,Citation10]. Seeded propagation of tau aggregates in vivo is thought to occur by cross-synaptic transfer of misfolded tau aggregates between neurons, prompting the misfolding of monomeric tau in the recipient neurons [Citation11–Citation13]. Pathological tau propagation via anatomically connected brain regions has thus been described as a ‘prion-like’ phenomenon [Citation14].
The prion-like propagation of tau filaments has been induced experimentally in cells and in laboratory animals. Intracranial inoculation of synthetic tau fibrils or brain materials containing tau aggregates is known to induce tauopathies in both wild-type and transgenic mice [Citation15–Citation17]. Notably, injection of brain lysates derived from various tauopathies resulted in the transmission of specific pathologies phenotypically reminiscent of the original human disorders, indicating tauopathies with prion-like strain characteristics [Citation18,Citation19]. The generation of conformationally distinct tau aggregates (or strains) has been described [Citation20,Citation21]. The degree of seeding activity in tau aggregates was reportedly influenced by aggregate size [Citation22,Citation23].
The conversion of monomeric soluble tau into detergent-insoluble aggregates is accompanied by the acquisition of partial protease resistance. In prions, the cellular prion protein (PrPC) refolds into a pathological conformation (PrPSc) [Citation24] and the acquisition of protease-resistance is universally employed to characterize PrPSc conformers [Citation25–Citation27]. Moreover, the size of protease-resistant core fragments representing distinct PrPSc conformers was maintained via in vivo transmissions or in vitro seeded conversions [Citation28–Citation30]. In contrast, the protease resistance characteristics of tau aggregates has not been thoroughly examined. In this study, we induced seeded tau aggregation in HEK293T cells using sarkosyl-insoluble fraction of AD brain tissue as exogenous seed, and compared the resulting proteolytic fragments of the tau aggregates generated in cultured cells with those from the AD brain material. The protease-resistant tau fragment newly formed in cultured cells was significantly lower molecular weight than the corresponding fragments from the parent seeds. Our results show that the in vitro formed tau species are conformationally distinct from original AD seeds.
Materials and methods
Human brain material
Human brain tissue was obtained from the Edinburgh Brain and Tissue Bank (EBTB) in Edinburgh, Scotland, UK. Frontal cortical samples derived from three patients with Alzheimer’s disease (AD) and three non-demented controls (Con) were used in this study (). Frozen brain samples pseudoanonymized using the Bank’s reference number system were sent to Korea Brain Research Institute (KBRI) without any patient identifiable data. Ethical approvals for the acquisition and use of human brain samples were obtained from KBRI’s Institutional Review Board. A piece of brain tissue (100–250 mg) was homogenized in nine volumes of phosphate-buffered saline (PBS, pH 7.4) containing complete EDTA-free protease inhibitors and phosphatase inhibitors (Roche Applied Science), by using the Precellys 24 tissue homogenizer (Bertin instrument). The 10% (w/v) homogenates were kept in aliquots at −80 °C until needed. The 10% homogenates were mixed with lysis buffer to a final concentration of 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100 and 0.5% NP-40. Homogenates were clarified by centrifugation at 1,000×g for 10 min at 4°C.
Table 1. Demographics of human cases used in this study.
Recovery of sarkosyl-insoluble fraction from human brains
A sarkosyl-insoluble fraction of human tau was recovered as described [Citation22,Citation31–Citation33] with minor modifications. Briefly, 350 μL of the 10% homogenates were mixed with the same volume of A68 buffer to a final concentration of 10 mM Tris-HCl, pH 7.4, 0.8 M NaCl, 1 mM EGTA, 5 mM EDTA, and 10% sucrose, followed by clarification at 13,000×g for 20 min. The supernatant was set aside and the pellets were resuspended in the same buffer and again subjected to centrifugation for 20 min. The supernatant portions from each centrifugation were combined, and sarkosyl was added to a final concentration of 1%. The mixture was incubated for 1 hour at room temperature on a flat rotating shaker set at 700 rpm, and subsequently centrifuged at 100,000×g for 1 hour at 4°C. The resultant pellets were suspended in PBS at 1/5th the starting volume of the 10% homogenates, and sonicated and stored at 4°C until use. This final sarkosyl-insoluble fraction of AD brains were used for the seeded polymerization of tau expressed in cultured cells (see below).
Stable cell lines
The HEK293T cells expressing tau repeat domain (tau RD) fused to enhanced green fluorescent protein (eGFP) were generated by WATSON RnD as described previously [Citation21,Citation34] with minor modifications. Briefly, a sequence encoding tau RD comprising amino acids 244–372 of the full-length 2N4R isoform of tau was inserted into pEGFP-N1 vector (Clontech Laboratories, Inc.) and then two point mutations (P301L and V337M) were generated by PCR mutagenesis. The resultant construct containing tau RD carrying P301L and V337M (hereinafter referred to as tau RD(LM)) fused to eGFP at the C-terminus through a 17-amino acid linker was obtained from the pEGFP-N1 vector using PCR amplification and then inserted into the XhoI and NotI sites of the pLVX-IRES-puro vector (Clontech Laboratories, Inc.).
HEK293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U of penicillin, 100 µg of streptomycin and 2 mM Glutamax in a humidified incubator at 37°C with 5% CO2. For packaging the viral particles, the cells were plated at a density of 5 × 105 cells in serum-free media in a 10 cm dish. On the following day, transfection was performed using 5 µg of the pLVX-tauRD, 3.4 µg of the pMDL g/pRRE, 1.7 µg of the pRSV-REV, 1.7 µg of the pVSV.G plasmid DNA and Gene-In transfection reagents (GlobalStem) according to the manufacturer’s instructions. Supernatants containing packaged lentiviral particles were harvested 48-hour later and concentrated using PEG-it (System Biosciences). HEK293T cells plated at a density of 5 × 104 cells per well in a 24-well-plate were transduced with the viral supernatants and selected in the media containing 2 µg/ml puromycin. Two monoclonal cell lines were isolated from the virally transduced polyclonal cells, and one of them was used as a cellular platform in this study to induce seeded aggregation.
Seeded tau aggregation assay in cultured cells
The HEK293T cells stably expressing tau RD(LM) fused to eGFP were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS, 100 U of penicillin and 100 µg of streptomycin, 2 mM Glutamax and 2 µg/ml puromycin in a humidified incubator at 37°C with 5% CO2. The sarkosyl-insoluble fraction of brain homogenates stored at 4°C were sonicated and used as inoculum to induce seeded tau aggregation in the cell culture. For seeded tau aggregation, the cells expressing tau RD(LM) were plated at a density of 2 × 105 cells per well on poly-D-lysine (PDL)-coated coverslips (Neuvitro Corporation) in a 12-well-plate followed by overnight incubation at 37°C. Inoculum was prepared by adding 10 µL of Lipofectamine 2000 and 40 µL of the sarkosyl-insoluble fraction to 350 µL of prewarmed Opti-MEM lacking FBS and antibiotics, followed by 20-min incubation at room temperature. After removal of the medium, the cells were overlaid with 400 µL of the prepared liposome mixture and incubated for ~20 hours at 37°C. Cells were removed from the plate by trypsin-EDTA treatment and passaged up to 20 times. While cells to be fixed for fluorescence microscopy were plated on the coverslips within wells of a 12-well-plate, those to be examined biochemically were plated in PDL-treated wells of a 6-well-plate. For normal passages, cells were cultured in 25 cm2 flasks without PDL treatment. For the preparation of cell lysates, cells removed from the plate were recovered by centrifugation at 2,000×g for 5 min at 4°C. Cell pellets to be assayed for the protease-resistant tau fragment were processed as described below. The pellets were lysed in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Triton X-100 and 0.5% NP-40, containing EDTA-free protease inhibitors (Roche Applied Science). Insoluble debris was removed by centrifugation at 2,000×g for 6 min at 4°C and supernatants were retained.
Proteolytic treatment
Enzymatic digestion of cell lysates was performed as described previously [Citation20] with minor modifications. Cells were recovered as described above and lysed in PBS containing 0.05% Triton X-100, then clarified by 5-min sequential centrifugations at 500×g and at 1,000×g, respectively. Total protein concentrations in the cell lysates were determined by performing a BCA assay (Pierce) and then adjusted with the lysis buffer to 4 mg/mL. The enzymatic digestion of sarkosyl-insoluble fraction of the brain homogenates was performed without adjustment of total protein content. For limited proteolysis, pronase prepared in PBS (1 mg/mL) was added to the cell lysates to a final concentration of 50 µg/mL and incubated for 1 hour at 37°C. The enzymatic reaction was terminated by the addition of sample buffer and subsequent incubation at 105°C, as described below.
Cell-free conversion of recombinant tau protein
Cell-free conversion of monomeric recombinant tau into an aggregated, protease-resistant form was conducted as described previously [Citation22] with some modifications [Citation21,Citation35]. The longest isoform of 2N4R recombinant tau obtained in the form of lyophilized powder (T-1001, rPeptide) was re-suspended with distilled water containing 16 mM dithiothreitol (DTT) immediately before use and allowed to stand at room temperature for 1 hour without shaking. Reaction mixtures (100 µL) in PBS plus 2.432 mM DTT contained recombinant tau protein as substrates at a concentration of 152 µg/ml and 5 µL of the sarkosyl-insoluble fraction of brain tissue as seeds. The mixtures were incubated at 37°C for 72 hours with cycles of 1 minute shaking (400 rpm) and 1 minute rest. The reaction products were digested with pronase (10 µg/mL) for one hour at 37°C. The enzymatic reaction was terminated by the addition of sample buffer and subsequent incubation at 105°C, as described below.
Fluorescence microscopy
For the fluorescent detection of tau aggregates, cells were fixed in fixation/solubilisation buffer (4% paraformaldehyde, 4% sucrose and 1% Tritox X-100 in PBS) for 15 min at room temperature. This stringent buffer condition was employed to remove soluble tau protein in cells [Citation36]. Where soluble tau observation is required, cells were first fixed in 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100. Following washes with PBS, fluorescent detection was acquired using a motorized fluorescent microscope (Nikon Ti-E equipped with NIS-Elements Imaging Software AR 4.40). Confocal images were obtained using a Nikon Ti-RCP confocal microscope (with a 60 × oil objective) coupled to NIS-C software system.
Antibodies
The following anti-tau antibodies were used in this study: RD4 (1:1,000, 05–804, Millipore), Tau5 (1:4,000, MA5-12808, ThermoFisher), 2B11 (1:500, 10237, IBL), AT8 (1:1,000, MN1020, ThermoFisher), AT100 (1:1,000, MN1060, Invitrogen), Ser262 (1:2,000, ab64193, Abcam), and pS396 (1:2,000, ab109390, Abcam). The mouse monoclonal antibody (mAb) RD4 (directed to amino acids 275–291 of 4-repeat [4R] tau) preferentially recognizes 4R isoform of tau [Citation37]. The mAbs 2B11 (directed to residues 306–312) and Tau5 (recognizing amino acids 218–225) react with both 3R and 4R isoforms of tau. The rabbit polyclonal serum Ser262 reacts with both non-phosphorylated and phosphorylated serine at residue 262. Phosphorylation-dependent antibodies AT8, AT100 and pS396 recognizes pSer202/pSer205, pThr212/pSer214/pThr217, and pSer396, respectively [Citation38,Citation39].
Tau ELISA
The concentrations of total tau and pSer396 tau in the sarkosyl-insoluble fraction from brains of human patients were measured using commercially available ELISA kits (KHB0041 for pSer396 tau and KHB7031 for total tau, Thermo Fisher Scientific), according to the manufacturer’s instructions.
Electrophoresis and Western blot analysis
Undigested or pronase-digested samples were mixed NuPAGE sample buffer (Invitrogen) to a final concentration of 1X (141 mM Tris base, 106 mM Tris HCl, 2% lithium dodecyl sulfate, 0.5 mM EDTA, 10% glycerol, 0.22 mM SERVA blue G250, and 0.175mM phenol red) containing 4% sodium dodecyl sulfate and 50 mM dithiothreitol. The mixed samples were incubated for 20 min in a heating block set at 105°C, and subsequent protein analysis was carried out using NuPAGE Novex gel systems (Invitrogen). Briefly, the proteins were run in either 10% or 4–12% Bis-Tris NuPAGE gels and were blotted on polyvinylidenedifluoride (PVDF) Immobilon-P membranes (Millipore). The membranes were blocked with 5% (w/v) dry milk in TBST (10 mM Tris-HCl, pH 8, 150 mM NaCl and 0.05% Tween-20) and tau was detected using the indicated primary antibodies. Subsequently, the membranes were probed either by 1:40,000 dilution of horseradish peroxidase-conjugated goat anti-mouse IgG F(ab’)2 fragments (Invitrogen) or by 1:100,000 dilution of horseradish peroxidase-conjugated donkey anti-rabbit IgG (GE Healthcare Life Sciences). Both primary and secondary antibodies were diluted in 1% (w/v) dry milk in TBST. The blots were then developed using ECL Prime reagent (GE Healthcare Life Sciences) and chemiluminescent detection was achieved using either Fusion FX7 SPECTRA (Vilber) or ChemidocTM XRS+ system (Bio-Rad). The molecular weight of tau protein fragments was determined by reference to SeeBlueTM Plus2 pre-stained protein standard (LC5925, Invitrogen) and IgG-binding MagicMarkTM XP Western protein standard (LC5602, Invitrogen).
Results
Immunoblot analysis of total tau and sarkosyl-insoluble tau in human brains
Brain lysates and sarkosyl-insoluble fractions were prepared from the frontal cortex of three AD cases and of three non-demented controls (). Tau profiles in these samples were explored by immunoblotting using the aforementioned antibodies (). When brain lysates were investigated using phosphorylation-independent anti-tau antibodies (Ser262, 2B11 and Tau5), tau profiles were similar between antibodies, characterized by full-length species migrating at 49–62 kDa and a group of truncated tau fragments less than 40 kDa () upper row; data not shown for Tau5). Notably, migration profiles of tau proteins were similar between AD cases and non-demented cases, although band intensities corresponding to full-length tau species were often weaker in AD cases than in the non-demented controls. We then examined tau profiles of the sarkosyl-insoluble fraction using the same set of phosphorylation-independent antibodies. As shown in the bottom row of ), immunoreactivities against tau were detectable only in AD3 but absent in the two other AD and the three non-demented control brains.
Figure 1. Immunoblot analysis of total tau and sarkosyl-insoluble tau in human brains. (a) Anti-tau antibodies used in this study were described with their recognition sites within the full-length 2N4R isoform of tau. (b) The brain lysate or sarkosyl-insoluble fraction from three cases of AD and three cases of non-demented controls were investigated by Western blot analysis with the use of phosphorylation-independent anti-tau antibodies Ser262 or 2B11. (c) The brain lysate or sarkosyl-insoluble fraction from the brains described in (b) were investigated by Western blot analysis with the use of phosphorylation-dependent anti-tau antibodies AT8 or AT100 or pS396. Molecular mass markers in kilodaltons are shown on the left.
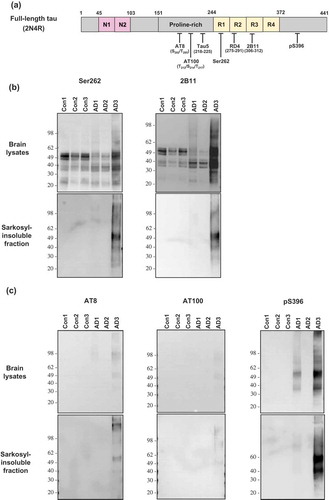
We next examined the brain lysate and the sarkosyl-insoluble fraction using phosphorylation-dependent antibodies, AT8, AT100 and pS396. As shown in ), immunoreactivities against AT8 and AT100 were undetectable both in the lysate and the sarkosyl-insoluble fraction of human brains except for AD3 in which faintly stained smears were barely visible. In comparison, the antibody pS396 strongly stained tau species and smears in the lysate and sarkosyl-insoluble fraction of AD3 brain (), right column); in other human brain samples, the anti-pS396 immunoreactivity was not detectable except for AD1 brain lysate in which faint tau bands were labelled. The measurement of tau species phosphorylated at residue 396 in the sarksoyl-insoluble fraction by using pS396 ELISA also showed results similar to those obtained by Western blot analysis (data not shown).
Collectively, tau profiles were quite similar between the lysates and the sarkosyl-insoluble fraction when investigated by the phosphorylation-dependent antibodies. When probed by the phosphorylation-independent antibodies, tau profiles obtained from sarkosyl-insoluble fraction were similar to those of phosphorylation-dependent antibodies. Among the six brain samples (three samples from AD patients and the other three from non-demented controls; all sampled from frontal cortex), sarkosyl-insoluble, aggregated and phosphorylated tau proteins were consistently detected only in the AD3 tissue.
HEK293T cells expressing tau RD(LM) fused to eGFP
The repeat domain of 4R isoform of human tau carrying two point mutations (P301L and V337M; tau RD[LM]) was fused to eGFP and then was expressed in HEK293T cells using a lentiviral transduction system for use as a model to examine prion-like tau aggregation [Citation21,Citation40]. Green fluorescent signals in the selected monoclonal HEK293T cell line were detectable in confocal microscopy (Figure 2(a)). Immunoblot analysis using anti-tau antibody RD4 labelled one band at ~45 kDa, indicating the expression of tau RD(LM)-eGFP ()). Western blots using an anti-eGFP antibody revealed the presence of an additional faint band at ~32 kDa, indicating that a minor portion of the expressed eGFP proteins may lack the C-terminal RD repeats ()). Additionally, one non-specific faint band at ~28 kDa was consistently detected with the anti-eGFP antibody in HEK 293T cells irrespective of protein expression.
Figure 2. Expression of tau RD(LM) fused to eGFP in HEK293T cells. Repeat domain (RD) of 4R tau mutated at two positions (P301L and V337M; tau RD[LM]) were fused to eGFP and expressed in HEK293T cells. A monoclonal HEK293T cell line stably expressing the mutant tau RD was selected and employed in this study. (a) A representative image of a monoclonal HEK293T cell line expressing tau RD(LM)-eGFP. For comparison, an image of HEK293T cells were shown in parallel. Scale bar = 50 µm. (b) The lysate of the monoclonal HEK293T cells carrying tau RD(LM)-eGFP was examined by Western blot analysis in comparison with that of normal HEK293T cells, with GAPDH as a loading control. The blots were probed using anti-tau antibody (RD4) or anti-eGFP antibody. Molecular mass markers in kilodaltons are shown on the left.
![Figure 2. Expression of tau RD(LM) fused to eGFP in HEK293T cells. Repeat domain (RD) of 4R tau mutated at two positions (P301L and V337M; tau RD[LM]) were fused to eGFP and expressed in HEK293T cells. A monoclonal HEK293T cell line stably expressing the mutant tau RD was selected and employed in this study. (a) A representative image of a monoclonal HEK293T cell line expressing tau RD(LM)-eGFP. For comparison, an image of HEK293T cells were shown in parallel. Scale bar = 50 µm. (b) The lysate of the monoclonal HEK293T cells carrying tau RD(LM)-eGFP was examined by Western blot analysis in comparison with that of normal HEK293T cells, with GAPDH as a loading control. The blots were probed using anti-tau antibody (RD4) or anti-eGFP antibody. Molecular mass markers in kilodaltons are shown on the left.](/cms/asset/dc492c52-68d2-4bcc-8715-7bafa422d5d3/kprn_a_1545524_f0002_oc.jpg)
Induction of seeded aggregation of tau RD(LM) in the cells and identification of protease-resistant core fragment
Previous studies have reported that seed-competent tau species from mouse tauopathy models are multimeric tau aggregates [Citation22,Citation41,Citation42]. We therefore used the sarkosyl-insoluble fraction obtained from two AD samples (AD2 and AD3; ) and two non-demented controls (Con1 and Con3) as exogenous seeds in the HEK293T cell expressing tau RD(LM)-eGFP. The cells were seeded for ~20 hours, passaged up to 20 times, and fixed at pre-determined passages in the condition in which soluble proteins were removed, and then visualized by fluorescent and confocal microscopy. While a majority of cells exposed to AD3 fluoresced with green aggregates, only very minimal green fluorescence was observed in cells treated with the non-demented controls (). Notably, some cells exposed to the sarkosyl-insoluble fraction of AD2 in which tau proteins were not detectable by Western blot featured with aggregates () middle row). As shown in ), numerous inclusions of various morphologies were observed in the majority of cells exposed to AD3 at passage 2; the proportion of cells containing green aggregates rapidly dropped in subsequent passages, but cells containing speckles were identified even at passage 20.
Figure 3. Identification of protease-resistant fragment of tau RD(LM) aggregates formed in HEK293T cells by seeded aggregation. The HEK293T cells expressing tau RD(LM) were exposed to the sarkosyl-insoluble fraction of brain samples for ~20 hours and passaged up to 20 times. (a) Cells at passage 2 were fixed in the condition to remove soluble proteins and the formation of tau RD(LM) aggregates were analysed by fluorescent microscopy. Scale bar = 100 µm. (b) Cells at multiple passages were fixed as in (a) and the formation of tau RD(LM) aggregates were examined by confocal microscopy. Scale bar = 50 µm. (c) Cells were recovered at different passages and lysed. Following the adjustment of total protein to 4 mg/mL, the lysates left undigested or digested with pronase at 50 µg/ml and examined by Western blot analysis using anti-tau antibody 2B11. Arrowheads indicate bands representing a monomeric form of pronase-resistant tau fragment. Arrows indicate bands representing a dimeric or trimeric form of proteolytic tau fragment. Molecular mass markers in kilodaltons are shown on the left. (d) The sarkosyl-insoluble fraction of AD3 or AD3-exposed cells recovered at passage 2 were digested with pronase at 50 µg/ml and then investigated by Western blot analysis using three anti-tau antibodies Ser262, 2B11 or RD4. Arrowheads indicate bands representing a monomeric form of pronase-resistant tau fragment. Arrows indicate bands representing a dimeric form of proteolytic tau fragment. Molecular mass markers in kilodaltons are shown on the left.
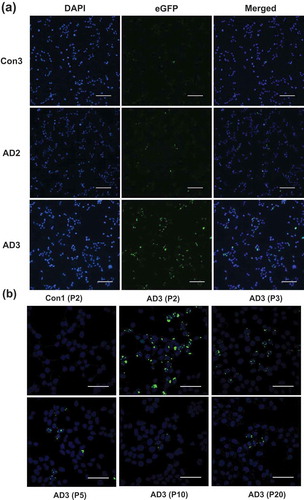
Figure 3. (Continued).
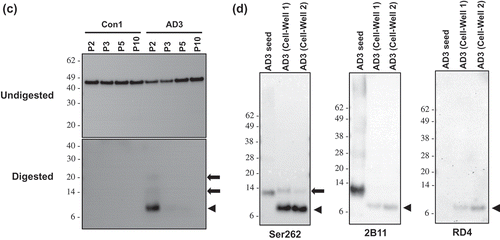
Similar to prions composed of PrPSc, aggregated forms of tau in AD yielded a protease-resistant fragment following digestion with pronase [Citation43]. Using pronase, we investigated whether in vitro produced tau RD(LM) aggregates also displayed protease-resistance. The cells exposed to the sarkosyl-resistant AD brain fractions were lysed at various passages, and either digested or not with pronase prior to immunoblotting. In all undigested samples, tau RD(LM)-eGFP was stained at ~45 kDa with similar intensities between them (), Undigested). No pronase-resistant tau was detectable for the cells seeded with Con1 (), Digested), however, the AD3-exposed cells collected at passage 2 produced a pronase-resistant tau fragment at ~7 kDa with two additional faint bands at ~14 kDa and ~21 kDa, likely representing dimers and trimers of the ~7 kDa fragment (), Digested).
The sarkosyl-insoluble fraction of the AD3 brain sample was then digested with pronase and its gel mobility was compared with the pronase-digested AD3 cell lysates at passage 2. The antibodies Ser262 and 2B11 detected a tau fragment migrating at 12–13 kDa with high-molecular-weight multimers in the digested AD3 sample ()). This 12–13 kDa tau species was previously described in studies examining the protease-protected core region of tau in AD brains [Citation43]. The pronase-resistant fragment from in vitro produced tau aggregates were 4–5 kDa lower molecular weight than AD seed fragments. ()). In contrast to antibodies Ser262 and 2B11, antibody RD4 did not recognize the 12–13 kDa AD-derived tau species while the cell-derived ~7 kDa fragment was revealed by RD4. This result is somewhat unexpected, given that RD4 epitope is located between those of Ser262 and 2B11. Nonetheless, this lack of immunoreactivity of 12–13 kDa tau species with the antibody RD4 may be explained by the post-translational deamidation occurring at residue 279 asparagine in AD brains [Citation44]. Collectively, the newly formed cellular tau RD aggregates were clearly distinguishable from the AD-derived seeds by their apparent molecular weight following proteolytic treatment as well as their reactivity with antibody RD4.
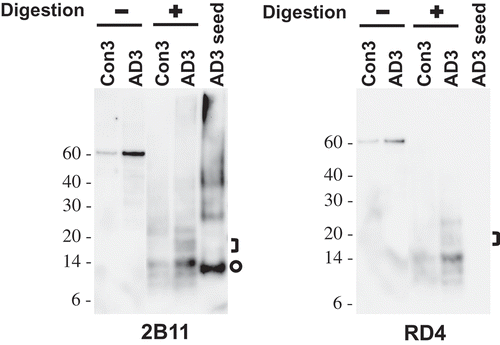
While the cell we used for the induction of seeded tau aggregation expressed a mutated fragment tagged with eGFP protein, a major species of tau in the sarkosyl-insoluble fraction of AD3 was largely full-length tau as shown in ). Since this intrinsic difference of tau between substrates and seeds might have led to the production of 4–5 kDa smaller proteolytic fragment, we investigated this possibility by employing cell-free conversion assay in which full-length recombinant tau was used as substrate. The reaction mixtures containing recombinant wild-type tau and the sarkosyl-insoluble fraction of brain homogenates were incubated at 37°C for 72 hours with intermittent shaking. The reaction products left undigested or digested with pronase prior to immunoblotting. In undigested samples, full-length tau protein was stained approximately at ~60 kDa (Figure 4). Following proteolytic treatment, several pronase-resistant tau fragments lower than 14 kDa or higher than 20 kDa were identified in both Con3- and AD3-seeded reactions suggesting that they were generated spontaneously irrespective of the presence of multimeric tau aggregates (). In contrast, bands migrating between 14 and 20 kDa were identified only in the reaction seeded with AD3 indicating that they were generated in a seed-dependent manner (); they were clearly distinguishable from 12-13 kDa tau species of AD3 seed. As described in the analysis of cell lysates, the antibody RD4 did not recognize the AD-derived proteolytic tau fragments at all.
Figure 4. Cell-free conversion of recombinant tau protein into the pronase-resistant form by seeded reaction. Recombinant tau proteins were mixed with the sarkosyl-insoluble fraction of brain samples and then incubated at 37°C for 72 hours with intermittent shaking (1 minute shaking at 400 rpm and 1 minute rest). The reaction products left undigested or digested with pronase at 10 µg/mL and examined by Western blot analysis using anti-tau antibodies 2B11 or RD4. The loaded amount of undigested sample was 1/5th of those digested. Pronase-digested AD3 seed was also run on the same gel for the comparison of gel mobility between seeds and in vitro (cell-free) generated species. Brackets designate bands generated in the reactions specifically seeded with AD3. Circles indicate bands representing a monomeric form of pronase-resistant tau fragment of the AD3 seed. Molecular mass markers in kilodaltons are shown on the left.
Discussion
Emerging evidence indicates that pathogenic tau assemblies can propagate and spread between synaptically connected neurons in a prion-like manner. Seeded conversion of soluble tau into insoluble aggregates is often accompanied by the acquisition of protease-resistance. In AD, pathogenic tau assemblies leave a 12 kDa pronase-resistant core fragment of 93–95 residues, largely composed of tau repeats [Citation43,Citation45]. Likewise, protease-resistant PrPSc is thought to retain strain-specific conformations that remain unaltered after in vivo transmission [Citation29,Citation30], in vitro cell propagation [Citation46] and during cell-free replication [Citation28]. Given the mechanistic similarities between the propagation of prions and filamentous tau assemblies, we hypothesized that the protease-resistant core tau would likewise persist during seeded aggregation. To test this hypothesis, we employed the HEK293T cell culture model expressing tauRD(LM)-fused to eGFP. Although this model has intrinsic limitations in studying trans-synaptic propagation of tau aggregates occurring in synaptically connected neurons, a group of similar cellular models have been widely employed in the field to understand molecular mechanisms underlying their uptake, propagation and strain characterization [Citation11,Citation12,Citation20–Citation22,Citation40,Citation41,Citation47,Citation48]. We first induced the seeded aggregation of tau RD(LM) in cultured cells following exposure to sarkosyl-insoluble tau from AD. Subsequently, the gel mobilities of protease-resistant tau fragments were analysed by immunoblot. Our data show that newly formed protease-resistant fragments were smaller than that of the parent AD seeds, thereby suggesting clear conformational differences between them.
Sanders and colleagues have shown that distinct patterns of proteolytic tau fragments from two tau prion strains can be maintained for multiple passages in cultured cells expressing tau RD(LM) fused to fluorescent protein [Citation21]. However, the direct comparison of protease-resistant fragment(s) between exogenous tau seeds and in vitro produced tau aggregates was not performed in the study. To the best of our knowledge, this study is the first to perform that comparison. Given the results from Sanders at al. [Citation21] and from in vitro studies of prion replication [Citation28,Citation46,Citation49], significantly different sizes of protease-resistant tau fragments between human brain aggregates and cell-derived aggregates were somewhat unexpected. The most likely explanation is that the two mutations (P301L and V337M) that were introduced into tau RD in order to increase the efficiency of seeded conversion [Citation21,Citation34] may have led to the production of lower molecular weight protease-resistant tau fragments relative to the AD seeds. This explanation is supported by various mutation-associated inherited human prion diseases (e.g. P102L and F198S) which are biochemically characterized by lower molecular weight proteolytic PrPSc fragments [Citation50–Citation52]. Given that proteolytic treatment of conformationally distinct PrPSc aggregates in prions may result in the production PrP fragment different in sizes [Citation25,Citation26], tauRD(LM) aggregates produced in the cell could be conformationally distinct from parent seeds of Alzheimer’s disease. Although it looks less likely to be the case, another possibility is that the cell-derived tauRD(LM) aggregates have different pronase susceptibility due predominantly to differences in primary structure rather than conformation.
An alternative explanation is that AD-derived sarkosyl-insoluble tau preparations are composed of multiple tau conformers, and that a minor conformer within this preparation might preferably seed the tau RD expressed in cells. While it is known that tau can adopt multiple self-propagating conformers with distinct biological properties [Citation18,Citation20], it is less well understood what the role of multiple tau isoforms might be in the conformational diversity of tau in AD brains [Citation53–Citation55]. Notably, a recent study has suggested that small amounts of 4R-only tau aggregates associated with age-related tau astrogliopathy in AD brains could selectively function as seeds in cultured cells expressing only RD(LM) of 4R tau [Citation40,Citation56]. Thus, a minor species of tau assembly, whose lower molecular-weight protease-resistant core was not detected by Western blot, might have selectively functioned as proteopathic seeds in our cell model expressing tau RD(LM) of 4R but lacking that of 3R.
One limitation this study has is that tau aggregates used as seeds were derived from one patient. Among brain samples from three AD patients we examined, sarkosyl-insoluble, aggregated and phosphorylated tau proteins were consistently identified in brain samples of only one patient AD3 (). While AD3 robustly induced seeded conversion of tauRD(LM) in the cell, as expected, AD2 lacking detectable tau aggregates in the sarkosyl-insoluble fraction induced barely visible green aggregates ()). The absence or minimal detection of pathogenic tau in brain samples of the two AD patients (AD1 and AD2) were unexpected, given that all three Alzheimer’s disease patients were pathologically confirmed. Since prefrontal cortex (Brodmann area [BA] 9) is known to be affected by AD in the late stage of disease progression [Citation57,Citation58], one possible explanation is that the brain area BA9 may not yet be affected by the disease in the two AD patients. Or it is also possible that the BA9 area is partly affected by the disease, but small amounts of tissue (100–250 mg) sampled for homogenization may not encompass affected part of the area. Since the source of aggregated tau is limited to one patient and isoform ratios in tau aggregates can vary depending on disease progression and/or brain regions [Citation59,Citation60], proteopathic tau aggregates obtained from more AD cases and other tauopathies need to be applied to the cellular system in order to determine whether our results can be generalized.
A recent study reported that cross-seeding of tau protein lead to the emergence of structural variant of fibrils [Citation61], again supporting the possibility that the difference in size we observed may be attributed to the mutated tau fragment tagged with eGFP. To explore this possibility, we have conducted cell-free conversion assay in which full-length recombinant tau was used as substrate. This cell-free assay generated AD-specific pronase-resistant tau species migrating between 14 and 20 kDa (), which is also different in size from that of parent tau seeds. It has to be mentioned that a technically similar cell-free conversion assay widely used in the field of prions also featured with protease-resistant PrP fragments different in size from those of parent PrPSc seeds [Citation62,Citation63]. Thus, our result in the cell-free conversion assay may have limitation in answering whether the size shift of proteolytic tau fragment in our cellular system is primarily due to the intrinsic difference of tau between substrates (mutated tau fragment fused to eGFP) and seeds (full-length tau species). It remains to be determined whether the same results will be obtained in seeded aggregation assay in the cell expressing other forms of tau protein (e.g. untagged full-length wild-type, untagged wild-type RD, etc.) or in transmission into laboratory animals.
Acknowledgments
The authors would like to thank Dr. Roger A. Moore for critical reading of the manuscript and Hyung-sup Jang for help with the figures.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378.
- Iqbal K, Grundke-Iqbal I, Zaidi T, et al. Defective brain microtubule assembly in Alzheimer’s disease. Lancet. 1986;2:421–426.
- Goedert M, Spillantini MG, Jakes R, et al. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526.
- Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. Embo J. 1990;9:4225–4230.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259.
- Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–622.
- Hof PR, Bouras C, Buee L, et al. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol. 1992;85:23–30.
- McKee AC, Kosik KS, Kowall NW. Neuritic pathology and dementia in Alzheimer’s disease. Ann Neurol. 1991;30:156–165.
- Saito Y, Ruberu NN, Sawabe M, et al. Staging of argyrophilic grains: an age-associated tauopathy. J Neuropathol Exp Neurol. 2004;63:911–918.
- McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43–64.
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852.
- Holmes BB, DeVos SL, Kfoury N, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–47.
- Liu L, Drouet V, Wu JW, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302.
- Clavaguera F, Tolnay M, Goedert M. The prion-like behavior of assembled tau in transgenic mice. Cold Spring Harb Perspect Med. 2017;7.
- Stancu IC, Vasconcelos B, Ris L, et al. Templated misfolding of Tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in Tau transgenic mice. Acta Neuropathol. 2015;129:875–894.
- Iba M, McBride JD, Guo JL, et al. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015;130:349–362.
- Guo JL, Narasimhan S, Changolkar L, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med. 2016;213:2635–2654.
- Boluda S, Iba M, Zhang B, et al. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129:221–237.
- Clavaguera F, Akatsu H, Fraser G, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–9540.
- Kaufman SK, Sanders DW, Thomas TL, et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron. 2016;92:796–812.
- Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–1288.
- Falcon B, Cavallini A, Angers R, et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J Biol Chem. 2015;290:1049–1065.
- Silveira JR, Raymond GJ, Hughson AG, et al. The most infectious prion protein particles. Nature. 2005;437:257–261.
- Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383.
- Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66:2096–2101.
- Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–778.
- Collinge J, Sidle KC, Meads J, et al. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–690.
- Castilla J, Morales R, Saa P, et al. Cell-free propagation of prion strains. Embo J. 2008;27:2557–2566.
- Scott MR, Will R, Ironside J, et al. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci U S A. 1999;96:15137–15142.
- Telling GC, Parchi P, DeArmond SJ, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082.
- Takahashi M, Miyata H, Kametani F, et al. Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 2015;129:895–907.
- Audouard E, Houben S, Masaracchia C, et al. High-molecular-weight paired helical filaments from alzheimer brain induces seeding of wild-type mouse tau into an argyrophilic 4R tau pathology in vivo. Am J Pathol. 2016;186:2709–2722.
- Santa-Maria I, Varghese M, Ksiezak-Reding H, et al. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J Biol Chem. 2012;287:20522–20533.
- Woerman AL, Stohr J, Aoyagi A, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A. 2015;112:E4949–58.
- Saijo E, Ghetti B, Zanusso G, et al. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. 2017;133:751–765.
- Vasconcelos B, Stancu IC, Buist A, et al. Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. 2016;131:549–569.
- de Silva R, Lashley T, Gibb G, et al. Pathological inclusion bodies in tauopathies contain distinct complements of tau with three or four microtubule-binding repeat domains as demonstrated by new specific monoclonal antibodies. Neuropathol Appl Neurobiol. 2003;29:288–302.
- Yoshida H, Goedert M. Sequential phosphorylation of tau protein by cAMP-dependent protein kinase and SAPK4/p38delta or JNK2 in the presence of heparin generates the AT100 epitope. J Neurochem. 2006;99:154–164.
- Porzig R, Singer D, Hoffmann R. Epitope mapping of mAbs AT8 and Tau5 directed against hyperphosphorylated regions of the human tau protein. Biochem Biophys Res Commun. 2007;358:644–649.
- Woerman AL, Aoyagi A, Patel S, et al. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci U S A. 2016;113:E8187–e96.
- Jackson SJ, Kerridge C, Cooper J, et al. Short fibrils constitute the major species of seed-competent tau in the brains of mice transgenic for human P301S tau. J Neurosci. 2016;36:762–772.
- Takeda S, Wegmann S, Cho H, et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat Commun. 2015;6:8490.
- Novak M, Kabat J, Wischik CM. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. Embo J. 1993;12:365–370.
- Dan A, Takahashi M, Masuda-Suzukake M, et al. Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer’s disease brain. Acta Neuropathol Commun. 2013;1:54.
- Jakes R, Novak M, Davison M, et al. Identification of 3- and 4-repeat tau isoforms within the PHF in Alzheimer’s disease. Embo J. 1991;10:2725–2729.
- Cronier S, Laude H, Peyrin JM. Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death. Proc Natl Acad Sci U S A. 2004;101:12271–12276.
- Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–15331.
- Kfoury N, Holmes BB, Jiang H, et al. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–19451.
- Nishida N, Harris DA, Vilette D, et al. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J Virol. 2000;74:320–325.
- Piccardo P, Dlouhy SR, Lievens PM, et al. Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57:979–988.
- Parchi P, Chen SG, Brown P, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L gerstmann-straussler-scheinker disease. Proc Natl Acad Sci U S A. 1998;95:8322–8327.
- Tagliavini F, Lievens PM, Tranchant C, et al. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Straussler-Scheinker disease A117V. J Biol Chem. 2001;276:6009–6015.
- Siddiqua A, Margittai M. Three- and four-repeat Tau coassemble into heterogeneous filaments: an implication for Alzheimer disease. J Biol Chem. 2010;285:37920–37926.
- Schmidt ML, Zhukareva V, Newell KL, et al. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 2001;101:518–524.
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917.
- Kovacs GG, Ferrer I, Grinberg LT, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 2016;131:87–102.
- Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404.
- Bouras C, Hof PR, Giannakopoulos P, et al. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex. 1994;4:138–150.
- Uematsu M, Nakamura A, Ebashi M, et al. Brainstem tau pathology in Alzheimer’s disease is characterized by increase of three repeat tau and independent of amyloid beta. Acta Neuropathol Commun. 2018;6:1.
- Hara M, Hirokawa K, Kamei S, et al. Isoform transition from four-repeat to three-repeat tau underlies dendrosomatic and regional progression of neurofibrillary pathology. Acta Neuropathol. 2013;125:565–579.
- Nizynski B, Nieznanska H, Dec R, et al. Amyloidogenic cross-seeding of Tau protein: transient emergence of structural variants of fibrils. PLoS One. 2018;13:e0201182.
- Orru CD, Wilham JM, Hughson AG, et al. Human variant Creutzfeldt-Jakob disease and sheep scrapie PrP(res) detection using seeded conversion of recombinant prion protein. Protein Eng Des Sel. 2009;22:515–521.
- Atarashi R, Wilham JM, Christensen L, et al. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods. 2008;5:211–212.