
ABSTRACT
Acute mountain sickness (AMS) can cause capillary hyper-permeability and vasogenic edema. However, its underlying mechanisms remained unclear and there is no previous in vitro study on AMS. We therefore conducted an in vitro study and examined whether continuous hypobaric hypoxia (CHH) could alter expression of junctional protein complex of vascular endothelial cells, causing hyper-permeabilization. EA.hy926 human endothelial cells were exposed to either CHH or normoxia for up to 24 h. Flow cytometry using annexin V/propidium iodide co-staining demonstrated that cell death had no significant difference at 12-h, but was increased by CHH at 24-h. Transendothelial resistance (TER) of endothelial cell monolayer was progressively decreased by CHH from 1-h to 24-h. Western blot analysis and immunofluorescence study demonstrated decreased expression levels of VE-cadherin, PECAM-1 and ZO-1 junctional proteins at both 12-h and 24-h exposure time-points. Interestingly, while the main form of ZO-1 (220 kDa) was decreased, its degraded form (100 kDa) was increased by 24-h CHH that might be linked to the increased cell death. Our data have demonstrated that CHH caused vascular endothelial hyper-permeability and defective junctional protein complex by reducing expression levels of VE-cadherin, PECAM-1, and ZO-1. Taken together, these data may explain pathophysiology underlying vascular hyper-permeability in AMS.
Introduction
The number of people, who have continuous hypobaric hypoxia (CHH), especially mountain climbers, miners, military personnel and astronomers who work at high-altitude, is increasing dramatically.Citation1,2 At above 3,000 m of height, more than 40–75% of mountain climbers will develop some degrees of acute mountain sickness (AMS), which ranges from mild to severe forms that may involve cerebral and pulmonary systems.Citation1,2 Hypoxemia caused by a high altitude can lead to hyper-perfusion, increased intracapillary pressure, and vascular leakage from circulatory systems within brain, lungs, as well as peripheral vessels.Citation3 One of the long-standing hypotheses for (patho)physiologic changes that cause vasogenic edema is capillary hyper-permeability induced by endothelial activation.Citation4
The vascular wall composes of endothelial cell lining, which controls fluid/electrolytes/solutes transport between intravascular and extravascular spaces.Citation5,6 An increase of paracellular transport is associated with pathological conditions and disorders.Citation7-9 Endothelial paracellular transport of fluid/electrolytes/solutes is normally controlled by intercellular junctional protein complexes, including adherens junction (AJ) and tight junction (TJ).Citation10,11 A number of proteins are within these junctional complexes, e.g. vascular endothelial (VE)-cadherin (for AJ) and zonula occludens-1 (ZO-1) (for TJ).Citation12,13 In addition, platelet endothelial cell adhesion molecule 1 (PECAM-1) is considered as an uncategorized essential junctional protein.Citation14
Isobaric hypoxia could lead to an increase in paracellular permeability of endothelial wall of an animal brain capillary mesh by unclear mechanisms.Citation15,16 A few lines of evidence have proposed redistribution, but not altered expression levels, of cytoskeletal proteins associated with ZO-1 to be involved in this defect.Citation17 Additional references have implicated the involvement of transcellular transport as the source of leakage in this type of hypoxia.Citation18,19 However, investigations on hypoxia-induced vascular leakage (both in vivo and in vitro) are limited. Moreover, whether CHH (which is different from isobaric hypoxia) affects vascular endothelial permeability has not been examined. The present study thus aimed to address alterations in expression of intercellular junctional protein complex, i.e. VE-cadherin, ZO-1, and PECAM-1 after endothelial cells were exposed to CHH.
Results and discussion
At sea level with an atmospheric pressure of 760 mmHg, the normal room air contains approximately 21% O2 with a partial pressure of oxygen (PO2) of approximately 159–160 mmHg. PO2 is reduced to around 100 mmHg in arterial blood circulation and < 30 mmHg inside the cells by cellular oxygen consumption to maintain normal cellular activities and metabolisms. With high-altitude exposure, PO2 in the air environment and in blood circulation is reduced based on the degree of hypobaric pressure. Our current study simulated high-altitude induced hypoxia by generating CHH environment within a hypoxic incubator chamber. Atmospheric pressure under CHH condition was set at 667 mmHg, which caused a reduction of PO2 to 140 mmHg inside the chamber throughout the present study. In parallel, the controlled cells were maintained at an atmospheric pressure of 757 mmHg with a PO2 of 159 mmHg throughout the study.
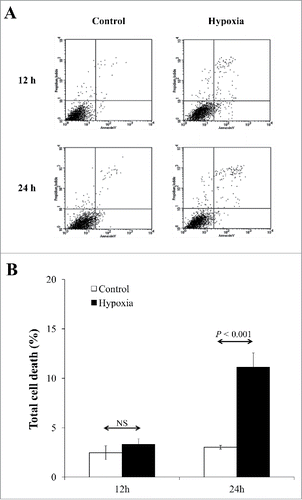
We exposed EA.hy926 human endothelial cells to CHH for up to 24 h, whereas those exposed to normoxia served as the controls. Microscopic examination showed no obvious differences in morphology between CHH and controlled cells. Flow cytometric analysis was performed to quantify cell death using annexin V/propidium iodide co-staining. The data showed that at 12-h exposure, cell death remained comparable between the 2 groups, whereas prolonged exposure to 24-h showed significant increase in cell death in CHH cells ().
Figure 1. Quantitative analysis of cell death. (A) Flow cytometric data using annexin V/propidium iodide co-staining. (B) Quantitative data obtained from 3 independent experiments. NS = not significant.
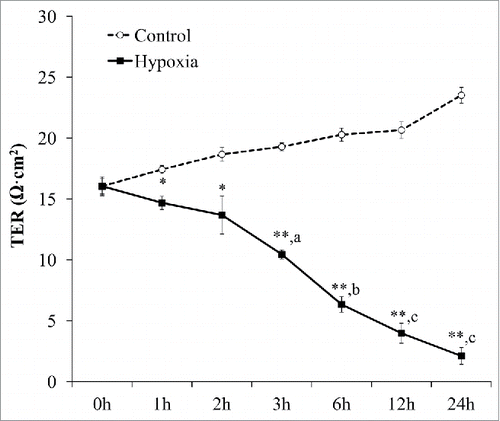
To assess whether these CHH affected permeability of vascular endothelial cell monolayer, TER was measured. Time-course measurements revealed that CHH markedly decreased TER at all-time points, as compared to basal TER of CHH cells and also to that of the controlled cells at all corresponding time-points (). Interestingly, while TER of the controls tended to increase, TER of CHH cells was progressively declined ().
Figure 2. Effects of CHH on vascular endothelial permeability. EA.hy926 cells grown in Traswell were exposed to CHH or normoxia for up to 24 h and transendothelial resistance (TER) was measured at various time-points. N = 3 independent experiments for each data point; * = p < 0.05 and ** = p < 0.01 vs. control at the matched time-point; a, b, and c = p < 0.005, p < 001, and p < 0.0005, respectively, vs. basal TER.
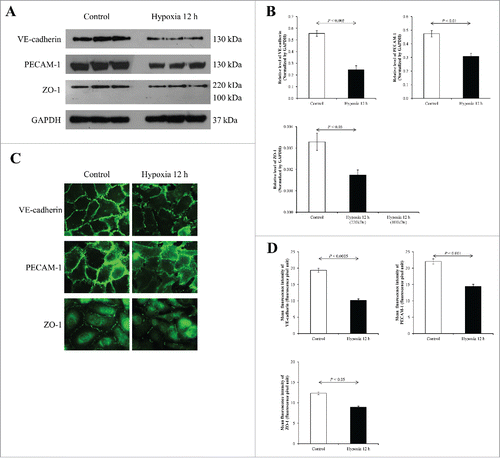
As aforementioned, junctional proteins play crucial roles in controlling vascular endothelial paracellular permeability, thereby, preventing leakage of fluid from one to the other sides. VE-cadherin (AJ protein), PECAM-1 (an uncategorized junctional protein), and ZO-1 (TJ-associated protein) were also evaluated for their expression levels in endothelial cells during CHH for 12 and 24 h. At 12-h, Western blot analysis of these proteins in EA.hy926 cells using responsible specific antibodies and normalized with GAPDH revealed decreased levels of VE-cadherin (130 kDa), PECAM-1 (130 kDa), and ZO-1 (220 kDa) (). Immunofluorescence study revealed decreases in expression levels of all these proteins in CHH cells, consistent to the Western blot data ().
Figure 3. Effects of 12-h CHH on VE-Cadherin, PECAM-1 and ZO-1 in EA.hy926 cells. (A) Western blotting of VE-Cadherin, PECAM-1 and ZO-1. (B) Quantitative band intensity using GAPDH as the loading control to normalize. N = 3 independent experiments. (C) Immunofluorescence staining of VE-Cadherin, PECAM-1 and ZO-1 (shown in green) in EA.hy926 cells. Original magnification = 200× for all panels. (D) The mean fluorescence intensity representing protein level was obtained from 10 random high power fields (HPFs) containing at least 100 cells for each conditions.
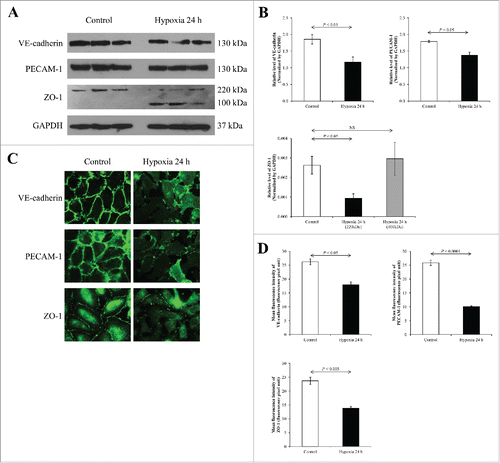
At 24-h exposure time, Western blot revealed decreased levels of VE-cadherin (130 kDa), PECAM-1 (130 kDa), and ZO-1 (220 kDa), similar to the data at 12-h time-point (). However, there was a newly present band of ZO-1 at approximately 100 kDa only in the CHH cells, most likely due to degradation of the main 220-kDa ZO-1 band. Immunofluorescence data showed marked decreases in expression levels of all these 3 junctional proteins, consistent to the Western blot data and the immunofluorescence data at 12-h ().
Figure 4. Effects of 24-h CHH on VE-Cadherin, PECAM-1 and ZO-1 in EA.hy926 cells. (A) Western blotting of VE-Cadherin, PECAM-1 and ZO-1. (B) Quantitative band intensity using GAPDH as the loading control to normalize. N = 3 independent experiments. (C) Immunofluorescence staining of VE-Cadherin, PECAM-1 and ZO-1 (shown in green) in EA.hy926 cells. Original magnification = 200× for all panels. (D) The mean fluorescence intensity representing protein level was obtained from 10 random high power fields (HPFs) containing at least 100 cells for each conditions.
Vascular leakage is a fatal clinical manifestations of AMS associated with CHH. To examine pathogenic mechanisms of vascular leakage induced by CHH, we focused our attention onto junctional protein complexes of EA.hy926 human endothelial cells, including VE-cadherin (an AJ protein), PECAM-1 (an uncategorized junctional protein), and ZO-1 (a TJ-associated protein).Citation12-14 These proteins have been previously investigated to define their roles in controlling paracellular permeability, particularly of the vascular wall. In a murine model, VE-cadherin knockout causes defective vascular remodeling, vascular collapse, and endothelial cell apoptosis.Citation20,21 Additional evidence has demonstrated that disruption of VE-cadherin solely is sufficient to increase vascular permeability.Citation22 ZO-1 is involved not only in linking the transmembrane TJ with intracellular cytoskeletal fibers but also in signal transduction to regulate TJ function.Citation23 ZO-1, which is a cytoplasm-linking part of TJ, is also expressed at the AJ due to its transient binding to β-catenin.Citation24 To regulate paracellular permeability, ZO-1 is activated by several phosphorylated amino acid residues.Citation25 Down-regulation of PECAM-1 is associated with defective vascular permeability in an animal model.Citation26 PECAM-1 is also associated with cadherin and several cytoplasmic proteins to modulate intercellular transport,Citation27 and with extracellular matrix-binding protein to control adhesive and migratory cellular activities.Citation28-30
Our present study has demonstrated effects of CHH on expression levels of junctional proteins and their function in vascular endothelial EA.hy926 cells. Because most important function of these junctional proteins is serving as a fence for soluble permeability, we monitored TER as an index of their function. The findings suggested that CHH markedly decreased TER in the time-dependent manner, which was in contrast to normoxia that progressively increased TER.
In summary, our findings showed that junctional proteins (VE-cadherin, PECAM-1 and ZO-1) were necessary for endothelial cell fence function as CHH reduced expression levels of these proteins to approximately half of the controlled levels. Defective permeability (or hyper-permeability) as reflected by the decreased TER shown in our present study was in concordance with the decreased expression levels of those 3 junctional proteins. Taken together, these findings may, at least in part, explain pathophysiology underlying vascular hyper-permeability in AMS.
Materials and methods
Cultivation of EA.hy926 cell line
EA.hy926 human endothelial cell line originated from the fusion of HUVEC with the lung carcinoma cell line A54922 was grown in D-MEM/F-12 medium (GIBCO; Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (GIBCO), 100 U/mL penicillin G and 100 mg/mL streptomycin (Sigma; St. Louis, MO). The cells were maintained in a humidified incubator with 21% O2 and 5% CO2 at 37°C.
Exposure to continuous hypobaric hypoxia (CHH)
For exposure to CHH, the cells were placed into a hypoxic incubator chamber (STEMCELL Technologies Inc.; Vancouver, Canada) (diameter 31 cm x height 11 cm), which was set at 37°C. The gas flow was removed from the chamber (at a rate of approximately 2 L/min) to reduce the atmospheric pressure to 667 mmHg. Parallel cells outside this chamber served as the controls. pH of the culture medium was maintained at 7.4 throughout the study.
Flow cytometric analysis of cell death using annexin V/propidium iodide co-staining
At 12 and 24 h after exposure to either normoxia or CHH, the cells were trypsinized with 3 mL of 0.1% trypsin in 2.5 mM EDTA and resuspended in 10 mL MEM. The harvested cells were centrifuged at 1,500 rpm and 4°C for 5 min, and washed with PBS. Cell pellets were resuspended with annexin V buffer (10 mM HEPES, 140 mM NaCl and 2.5 mM CaCl2.2H2O; pH 7.4) at a final concentration of 5 × 105 cells/mL and then incubated with FITC-labeled annexin V (BD Biosciences; San Jose, CA) on ice for 15 min in the dark. Propidium iodide (BD Biosciences) at the final concentration of 0.2 µg/mL was added to the cell suspension prior to analysis by flow cytometry. The cells were then analyzed by flow cytometry (FACScan, Becton Dickinson Immunocytometry System; San Jose, CA).
Measurement of transendothelial resistance (TER)
TER was measured after EA.hy926 cells were exposed to either CHH or normoxia for 0, 1, 2, 3, 6, 12 and 24 h using a Millicell-ERS resistance system (Millipore; Bedford, MA) following the manufacturer's instructions. TER was measured at 3 different sites in each Transwell and the resistance of polycarbonate membrane was subtracted.
Western blot analysis
Whole cells were resuspended in Laemmli's buffer and the recovered proteins were measured by Bradford's method using Bio-Rad Protein Assay (Bio-Rad Laboratories; Hercules, CA). An equal amount of total protein (30 mg) was resolved in each lane of 10% SDS-PAGE and the separated proteins were transferred onto a nitrocellulose membrane. Non-specific bindings were blocked with 5% skimmed milk in PBS at RT for 1 h, and the membrane was incubated with mouse monoclonal anti-VE-cadherin (Santa Cruz Biotechnology; Santa Cruz, CA), anti-PECAM-1 (Santa Cruz Biotechnology), anti-ZO-1 (Zymed Laboratories Inc.; San Francisco, CA), or anti-GAPDH (Santa Cruz Biotechnology) (as the loading control) antibody. All primary antibodies were diluted 1:1,000 in 1% skim milk/PBS. After washing with PBS 3 times, the membrane was then incubated with corresponding secondary antibody conjugated with horseradish peroxidase (Dako; Glostrup, Demark) (1:2,000 in 1% skim milk/PBS) at RT for 1 h. Immunoreactive bands were detected by SuperSignal West Pico chemiluminescence substrate (Pierce Biotechnology; Rockford, IL) and autoradiogram. Quantification of band intensity was performed using ImageMaster 2D Platinum software (GE healthcare; Uppsala, Sweden).
Indirect immunofluorescence study
EA.hy926 cell monolayers were grown on coverslip 1 day prior to CHH exposure. After 12 and 24 h exposure to CHH or normoxia (control), the cells were rinsed with PBS and fixed/permeabilized with ice-old methanol for 3 min at −20°C. After washing with PBS, cells were blocked with 1%BSA/PBS for 1 h at RT. Cells were incubated with mouse monoclonal antibody to VE-cadherin (Santa Cruz Biotechnology), PECAM-1 (Santa Cruz Biotechnology), or ZO-1 (Zymed Laboratories Inc.), all with a dilution of 1:100 in 1%BSA/PBS at 4°C overnight. The cells were then incubated with AlexaFluor-488-conjugated goat anti-mouse IgG (Invitrogen/Molecular Probes; Burlington, Canada) (1:5,000 in 1% skim milk/PBS) at RT for 1 h. Unbound secondary antibody was removed by rinsing with PBS and coverslips were mounted with 50% glycerol/PBS. Images were taken under a laser scanning microscope (LSM 510 Meta, Carl Zeiss; Jena, Germany). The quantitative data (mean fluorescence intensity) was obtained from 10 random high-power fields (HPFs) containing at least 100 cells for each condition using ImageJ software (version 1.48b) (http://imagej.nih.gov/ij/).
Statistical analysis
All quantitative data are presented as mean ± SEM. The unpaired Student's t-test was used for comparisons between 2 sets of the samples. P-value less than 0.05 was considered statistically significant.
Abbreviations
| AJ | = | adherens junction |
| AMS | = | acute mountain sickness |
| CHH | = | continuous hypobaric hypoxia |
| PECAM-1 | = | platelet endothelial cell adhesion molecule 1 |
| TER | = | transendothelial resistance |
| TJ | = | tight junction |
| VE-cadherin | = | vascular endothelial cadherin |
| ZO-1 | = | zonula occludens-1 |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
DS, PP and VT designed research; DS and PP performed experiments; DS, PP and VT analyzed data; DS and VT wrote the manuscript; All authors reviewed the manuscript.
Funding
This study was supported by Mahidol University research grant, Office of the Higher Education Commission and Mahidol University under the National Research Universities Initiative, and the Thailand Research Fund (RTA5680004). VT is also supported by “Chalermphrakiat” and “Research Staff” grants by Faculty of Medicine Siriraj Hospital.
References
- Wright AD. Medicine at high altitude. Clin Med (Lond) 2006; 6:604-8; PMID:17228562; https://doi.org/10.7861/clinmedicine.6-6-604
- Hackett PH, Rennie D, Levine HD. The incidence, importance, and prophylaxis of acute mountain sickness. Lancet 1976; 2:1149-55; PMID:62991; https://doi.org/10.1016/S0140-6736(76)91677-9
- Hackett PH. High altitude cerebral edema and acute mountain sickness. A pathophysiology update. Adv Exp Med Biol 1999; 474:23-45; PMID:10634991; https://doi.org/10.1007/978-1-4615-4711-2_2
- Hackett PH, Yarnell PR, Hill R, Reynard K, Heit J, McCormick J. High-altitude cerebral edema evaluated with magnetic resonance imaging: clinical correlation and pathophysiology. JAMA 1998; 280:1920-5; PMID:9851477; https://doi.org/10.1001/jama.280.22.1920
- Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 2004; 84:869-901; PMID:15269339; https://doi.org/10.1152/physrev.00035.2003
- Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci 2008; 1123:134-45; PMID:18375586; https://doi.org/10.1196/annals.1420.016
- Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008; 11:109-19; PMID:18293091; https://doi.org/10.1007/s10456-008-9099-z
- Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, Hartnett ME. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res 2008; 27:331-71; PMID:18653375; https://doi.org/10.1016/j.preteyeres.2008.05.001
- Weis SM. Vascular permeability in cardiovascular disease and cancer. Curr Opin Hematol 2008; 15:243-9; PMID:18391792; https://doi.org/10.1097/MOH.0b013e3282f97d86
- Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta 2008; 1778:794-809; PMID:17961505; https://doi.org/10.1016/j.bbamem.2007.09.003
- Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 2008; 121:2115-22; PMID:18565824; https://doi.org/10.1242/jcs.017897
- Cai J, Wu L, Qi X, Li CS, Caballero S, Shaw L, Ruan Q, Grant MB, Boulton ME. PEDF regulates vascular permeability by a gamma-secretase-mediated pathway. PLoS One 2011; 6:e21164; PMID:21695048; https://doi.org/10.1371/journal.pone.0021164
- Bauer H, Zweimueller-Mayer J, Steinbacher P, Lametschwandtner A, Bauer HC. The dual role of zonula occludens (ZO) proteins. J Biomed Biotechnol 2010; 2010:402593; PMID:20224657; https://doi.org/10.1155/2010/402593
- Privratsky JR, Newman PJ. PECAM-1: regulator of endothelial junctional integrity. Cell Tissue Res 2014; 355:607-19; PMID:24435645; https://doi.org/10.1007/s00441-013-1779-3
- Josko J, Knefel K. The role of vascular endothelial growth factor in cerebral oedema formation. Folia Neuropathol 2003; 41:161-6; PMID:14604298
- Fischer S, Clauss M, Wiesnet M, Renz D, Schaper W, Karliczek GF. Hypoxia induces permeability in brain microvessel endothelial cells via VEGF and NO. Am J Physiol 1999; 276:C812-C820; PMID:10199811
- Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol 2002; 282:H1485-H1494; PMID:11893586; https://doi.org/10.1152/ajpheart.00645.2001
- Plateel M, Teissier E, Cecchelli R. Hypoxia dramatically increases the nonspecific transport of blood-borne proteins to the brain. J Neurochem 1997; 68:874-7; PMID:9003080; https://doi.org/10.1046/j.1471-4159.1997.68020874.x
- Cipolla MJ, Crete R, Vitullo L, Rix RD. Transcellular transport as a mechanism of blood-brain barrier disruption during stroke. Front Biosci 2004; 9:777-85; PMID:14766407; https://doi.org/10.2741/1282
- Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999; 98:147-57; PMID:10428027; https://doi.org/10.1016/S0092-8674(00)81010-7
- Gory-Faure S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M, Huber P. Role of vascular endothelial-cadherin in vascular morphogenesis. Development 1999; 126:2093-102; PMID:10207135
- Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro A, Ruco L, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A 1999; 96:9815-20; PMID:10449777; https://doi.org/10.1073/pnas.96.17.9815
- Gonzalez-Mariscal L, Betanzos A, Avila-Flores A. MAGUK proteins: structure and role in the tight junction. Semin Cell Dev Biol 2000; 11:315-24; PMID:10966866; https://doi.org/10.1006/scdb.2000.0178
- Itoh M, Nagafuchi A, Moroi S, Tsukita S. Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments. J Cell Biol 1997; 138:181-92; PMID:9214391; https://doi.org/10.1083/jcb.138.1.181
- Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 1999; 294:1351-62; PMID:10600390; https://doi.org/10.1006/jmbi.1999.3310
- Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest 2002; 109:383-92; PMID:11827998; https://doi.org/10.1172/JCI0213595
- Sheibani N, Sorenson CM, Frazier WA. Differential modulation of cadherin-mediated cell-cell adhesion by platelet endothelial cell adhesion molecule-1 isoforms through activation of extracellular regulated kinases. Mol Biol Cell 2000; 11:2793-802; PMID:10930470; https://doi.org/10.1091/mbc.11.8.2793
- Jackson DE, Kupcho KR, Newman PJ. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of platelet/endothelial cell adhesion molecule-1 (PECAM-1) that are required for the cellular association and activation of the protein-tyrosine phosphatase, SHP-2. J Biol Chem 1997; 272:24868-75; PMID:9312087; https://doi.org/10.1074/jbc.272.40.24868
- Sun QH, DeLisser HM, Zukowski MM, Paddock C, Albelda SM, Newman PJ. Individually distinct Ig homology domains in PECAM-1 regulate homophilic binding and modulate receptor affinity. J Biol Chem 1996; 271:11090-8; PMID:8626652; https://doi.org/10.1074/jbc.271.19.11090
- Tanaka Y, Albelda SM, Horgan KJ, van Seventer GA, Shimizu Y, Newman W, Hallam J, Newman PJ, Buck CA, Shaw S. CD31 expressed on distinctive T cell subsets is a preferential amplifier of beta 1 integrin-mediated adhesion. J Exp Med 1992; 176:245-53; PMID:1377224; https://doi.org/10.1084/jem.176.1.245