
ABSTRACT
Angiogenesis plays an important role in controlling tissue development and maintaining normal tissue function. Dysregulated angiogenesis is implicated in the pathogenesis of a variety of diseases, particularly diabetes, cancers, and neurodegenerative disorders. As the major regulator of angiogenesis, the vascular endothelial growth factor (VEGF) family is composed of a group of crucial members including VEGF-B. While the physiological roles of VEGF-B remain debatable, increasing evidence suggests that this protein is able to protect certain type of cells from apoptosis under pathological conditions. More importantly, recent studies reveal that VEGF-B is involved in lipid transport and energy metabolism, implicating this protein in obesity, diabetes and related metabolic complications. This article summarizes the current knowledge and understanding of VEGF-B in physiology and pathology, and shed light on the therapeutic potential of this crucial protein.
Introduction
Angiogenesis is highly coordinated by a series of orchestrated events, and the interactions of VEGF family to its receptor have been well characterized. The VEGF family is major regulators of blood and lymphatic vessel development and growth,Citation1 comprising VEGF-A, -B, -C, -D, placenta growth factor (PlGF). In some literatures the VEGF-E (an Orf-virus-encoded protein) and VEGF-F (a variant isolated from snake venom)Citation2 were also included in this family. These grow factors are secreted as ∼40-kDa dimeric glycoproteins and function in a paracrine fashion by signaling to three corresponding structurally homologous receptor tyrosine kinases expressed on endothelium cells (ECs) – vascular endothelial growth factor receptor (VEGFR)-1, −2 −3.Citation3 VEGF-A, VEGF-B and PlGF also bind to the neuropilin (NRP)-1 and −2.Citation4 Most biological angiogenic event occur through the VEGFR-2, whereas VEGFR-1 acts by synergistically augmenting VEGFR-2 signaling.Citation2 Their binding patterns are partially overlapping, and the feasible cross-talks may amplify the diversification of intracellular and intercellular interchange of communication. For example, the PlGF strengthened the activity of VEGF-A by displacing VEGF-A from VEGFR-1, facilitating its availability to the VEGFR-2.Citation5 Conversely, the transduced VEGF-B in RIP1-Tag2 islets possibly replaced VEGF-A and PlGF from VEGFR-1, and then diminishing pro-angiogenic effect.Citation6 In addition, the VEGFs are also engaged in the conversion from white adipose tissue (WAT) to brown adipose tissue (BAT),Citation7 leading to increased energy expenditure, and resulting in protection from diet induced obesity. The coexistence of angiogenic and browning effect may coordinate the organism to obtain a better adaptation to the external.
Discovered in 1996,Citation8 VEGF-B has approximate 47% and 37% amino acids sequence identical with VEGF-A and PlGF.Citation9 Owing to alternative splicing event, the VEGF-B gene generates two isoforms: VEGF-B167 and VEGF-B186, 42/60 KDa homodimers, respectively.Citation9 Their N-terminal contains the receptor binding domain,Citation10 homologous with the regions in VEGF-A and PlGF, therefore sharing the common receptors. The diversity in their C-terminal properties affects their distribution in the body. VEGF-B167 has a heparin-binding domain, thus upon exudation it binds to cell surface heparin sulfate proteoglycans to anchor this isoform in extracellular matrix.Citation8 Unlike VEGF-B167, VEGF-B186 does not connect the heparin, hence more diffusible.Citation11 The ratio of VEGF-B167/VEGF-B186 varies significantly among species. The VEGF-B167 is prevalent in mouse, while the human tumor cell lines favor the VEGF-B186.Citation12 Physiologically, the VEGF-B covers manifold organs and tissues, with the highest in the heart, skeletal muscle and lower levels in other tissues in adult mice.Citation13,Citation14
The in vivo role of VEGF-B remained elusive for decades. Due to its homologous structures, VEGF-B was initially recognized as an angiogenic factor. Subsequent studies, however, argued against the angiogenic activity of this molecule.Citation15-Citation18 Physiologically, VEGF-B has little growth effects, as demonstrated in gain-of-function studies using transgenicCitation15 and adenoviralCitation16 expression of VEGF-B models and loss-of-function studies using VEGF-B null mice.Citation17,Citation18 Under pathological conditions, this molecule can prevent cells from apoptosis and death. It showed both survival effect in laser injury-induced choroidal neovascularization or ischemia-originated retinal neovascularization models,Citation19 cardiac ischemia mouse,Citation20 and neuron-protective effect for the brain cortical neurons and retinal neuronsCitation21 and motor neurons in the spinal cord.Citation22 The two effects may be relatively complemented, since the neural and vascular systems are inseparable and share the common molecular mechanisms for migration.Citation23 To underlie the survival effects, besides the anti-apoptotic effect via repressing the expression of pro-apoptotic BH3-only proteins and other apoptosis- and cell death-related proteins, including p53 and caspase family members,Citation21 VEGF-B might potentially enhance energy metabolism by regulating fatty acid (FAs) transport.Citation9,Citation13 Surprisingly, at the high levels, VEGF-B acted as a growth-inhibiting molecule to forestall overgrowth and tumor growth.Citation6,Citation9
Collectively, VEGF-B is more like a survival molecule rather than a growth factor.Citation24 Recently its participation in lipid transport and energy metabolism mediation was partially revealed, indicating its implication in lipids accumulation relevant metabolic diseases, e.g. the type 2 diabetes mellitus (T2DM). Here, we summarized recent advances on VEGF-B studies, with particular interest on its potential therapeutic application in diabetes therapy.
Diabetes, from lipid depots to targeting VEGF-B therapy
The prevalence of diabetes has been increasing during the past decades and, more importantly, diabetes is associated with a variety of severe complications, particularly cardiovascular events and renal dysfunction.Citation25 T2DM is characterized as insufficient insulin secretion from pancreatic β-cell, impaired insulin-stimulated glucose uptake into skeletal muscle and adipose tissue and defective insulin-dependent suppression of hepatic glucose output,Citation26 and these defects collectively result in hyperglycemia. Insulin resistance, a state that the metabolically active cells are less sensitive to the insulin dependent glucose handing, marks and exacerbates the T2DM. Insulin resistance stimulates β-cells to augment insulin production to lower blood glucose, and the compensate degree determines whether the individual develops diabetes or not.Citation27 Both genic mutations and environmental factors can lead to the T2DM, and the latter may contribute more, such as the obesity. Obesity, via deteriorating insulin resistance, places an depraved functional demand on the β-cell and accelerate β-cell failure.Citation25
The classical remedy for T2DM is stepwise, starting from life style interventions such as caloric restriction via a very low-calorie diet (600 kcal/day)Citation28 or the exercise training;Citation29 afterwards the oral monotherapy, including incretion-based therapies such as glucagon-like peptide-1 and its stable analogs,Citation30 oral hypoglycemic agents (metformin and thiazolidinediones); further combination therapy, and ultimately insulin therapy.Citation31 In the following text, we first stated how the lipid dysregualtion contributes to insulin resistance development, then the targeted insulin resistance and VEGF-B for diabetes therapy.
Lipids metabolism, insulin resistance and T2DM
The relevance between lipid dysregulation and insulin resistance has been widely studied. Initially the Randle hypothesis explained that accumulative fatty acids (FAs) impaired pyruvate dehydrogenase and glycolysis.Citation32 Nevertheless, the diacylglycerol hypothesisCitation33 explicated the impaired insulin action and glucose disposal in chronic obesity states. Studies demonstrated the diacylglycerol and ceramides as pathogenic factors for insulin resistance.Citation34 The accumulation of them deteriorate insulin resistance by activating protein kinase C (PKC) family membersCitation35 and impairing the Akt2 action,Citation36 separately. Mechanistically, PKC-θ phosphorylates insulin-receptor substrate (IRS-1) on ser 1101 to block IRS-1 tyrosine phosphorylation;Citation35 and ceramides impedes Akt2 activation via protein phosphatase 2A dephosphorylating,Citation36 further disrupting insulin signaling. Importantly, it is the intramyocellular diacylglycerols,Citation37 not the circulating lipids, that interrupt the insulin signaling and are responsible for insulin resistance progression. Besides, ectopic deposition of lipids in metabolically active organs can induce pathological activation of inflammationCitation38,Citation39 and endoplasmic reticulum stress,Citation38,Citation40 the ability of which to regulate insulin action may be reliant on their ability to alter the levels of key signaling intermediates.Citation38 It is highly likely that dysregulations of these pathways collectively contribute to insulin resistance.
Indeed, recent studies suggest that it is the lipids accumulation in liver and muscle, but not in subcutaneous or visceral adiposity, accounts for insulin resistance.Citation41 The lipids in the islets are also concerned. Free fatty acids (FFAs) promote insulin production in the short termCitation42 yet repressing by interfering the co-localization of calcium channels and the secretory granulesCitation43,Citation44 in the long term. Obesity and high fatty diet (HFD) duplicated the effects of long-term incubation of islets with FFAs on the Ca2+ channel distribution and insulin output.Citation45 To figure out, the damaged β-cell function, rather than the declined number, may be the nature for the impaired insulin exudation.Citation25 generalized the pathological progression from ectopic lipids deposition to insulin resistance and T2DM development. To conclude, the ectopic lipids aggradation is vital, and normalization of lipid storage might be able to attenuate insulin resistance in metabolically active organs such as liver and muscle.
Figure 1. The progression from the ectopic lipids depots to the T2DM. Once the lipids overwhelm the capacity of adipose tissues, it shunts to the non-adipose tissues, leading to ectopic lipid deposition. (1), (2). The lipids deposition in the liver/muscle arouse the abnormal insulin behavior, resulting in muscle/hepatic insulin resistance. The insufficient insulin action gives rise to the glucose release from the liver and the lipids release from the adipose. Whereas, the glucose uptake is limited, relative to an increased lipids uptake by the tissue cells. (3). In the pancreatic islets, the lipid deposition would result in β-cell dysfunction and apoptosis,Citation6,Citation46 and the weaken insulin production. (4). The lipids depots on the artery intima lead to the coronary atherosclerotic disease, and this can further develop into latter ischemic heart disease, eventually the heart failure (HF). (5), (6). The muscle/hepatic insulin resistance repress the glucose uptake. To let down the glucose level, the β-cell produces more insulin, which does not work for the already existing insulin resistance. The functional adaptation of the β-cell bring about a high rates of β-cell metabolism and risk of β-cell damage from mitochondrial and endoplasmic reticulum stress.Citation29 (7). Insulin resistance would impair storage of carbohydrate as glycogen in muscle, then carbohydrates are redirected to the liver and become substrates for hepatic de novo lipogenesis.Citation47 (8). The hepatic insulin resistance can deteriorate into the non-alcoholic fatty liver disease (NAFLD), even the more severe non-alcoholic steatohepatitis, or hepatocellular carcinoma (HCC). (9) – (11). These combinations together to cause the final T2DM. (12). The NAFLD and T2DM are regularly co-existing. The NAFLD imposes the risk for the diabetes and its complications, in turn, diabetes makes an individual more likely to have more severe NAFLD with the associative complications of cirrhosis and mortality.Citation48
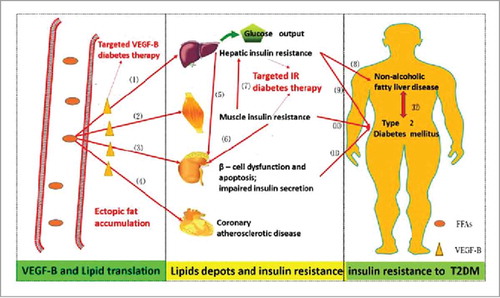
Figure 2. The involvement of the VEGF-B in the lipids translation, and the observed phenotype in the targeted VEGF-B treatment in various rodent animals. The black line stands for the pathways under physical conditions; the red for the pathological conditions; the green for the changes after the neutralizing VEGF-B strategies (the VEGF-B−/− model or VEGF-B antibody treatment). Under normal conditions, the PGC1-α regulates the coexpression of VEGF-B and the lipids oxidation associated genes, thus establishing a balance between the VEGF-B medicated lipids uptake and the energy demands of the metabolic cells, and the excessive lipids are stored in the adipocytes. When pathologic, the redundant lipids in the non-adipocyte tissues cause the abnormal insulin behavior, inducing the subsequent insulin resistance and the T2DM. Genetic or pharmacological inhibition of VEGF-B signaling leads to the (1). Decreased distribution of FATP3/4 on the ECs; (2). Less intracellular lipid droplets in working tissues; promoted insulin sensitivity and glucose uptake, ameliorated glucose tolerance; metabolic transformation from FAs to glucose oxidation; a lower risk for CVD; protected islet architecture and β-cell apoptosis; (3). Lipids redistribution to adipose tissues, leading to weight gain.
Treating diabetes by targeting insulin resistance
The peroxisome proliferator-activated receptors (PPARs) work as lipid sensors, modulating metabolic events by coordinately regulating the expression of genes linked to the energy homeostasis and insulin action, and therefore it can be regarded as a pharmacological target for management of metabolic disorders.Citation49 Thiazolidinediones (TZDs) can directly reduce peripheral systemic insulin resistance,Citation50 via the mighty activation of PPARγ, inducing the fat redirection from visceral to subcutaneous depots.Citation51 Given adipocytes own the highest PPARγ levels, these cells are the primary target for the glucose-lowering actions of TZDs.Citation52 Another insulin sensitizer-the apelin is also concerned with the magnified phosphorylation of Akt and glucose uptake in skeletal muscle.Citation53
Hurdles remain since most TZDs exert the risk of cardiovascular morbidities, and rosiglitazone has been withdrawn from the market.Citation54 To solve this issue, the selective peroxisome proliferator-activated receptor estrogen receptor modulatorsCitation55 might provide a more tolerable therapy for T2DM, without the cardiomegaly adverse effect or fewer. Additionally, the dual agonists of PPAR-α/γ or even PPAR-α/γ/δ pan agonistsCitation56 showed promising results in the simultaneous treatment of diabetic hyperglycemia and dyslipidemia.
VEGF-B and lipids transportation
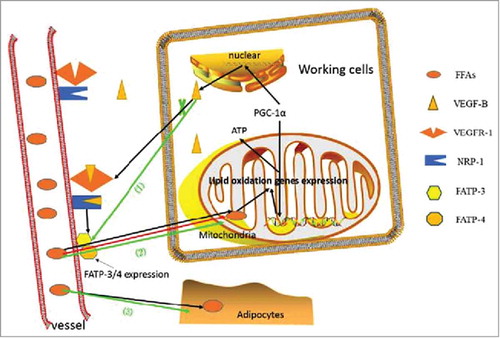
The VEGF-B is critical in coordinating ECs-mediated long-chain fatty acids (LCFA) uptake with the energy demand of the surrounding tissue via its co-expression with the mitochondrial gene cluster,Citation13 consisting primarily of genes coding for proteins within the oxidative phosphorylation machinery.Citation57 This may be under the transcriptional regulation of estrogen-related receptor α and co-regulator peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α).Citation58 VEGF-B released by the tissue cells promotes the distribution of fatty acids transport proteins (FATP)3 and FATP4 on the ECs, via its binding to VEGFR-1 and NRP-1, further facilitating the lipids transport into the tissue cells. The receptor knockout studies abolished the growing expression of FATP3/4Citation13 while the co-expression of two FATPs led to the highest uptake of LCFA, suggesting a synergistic effect. Both isoforms of VEGF-B promoted the expression of FATP3/4 in several lipid-metabolizing peripheral tissues at transcriptional and translational levels, with the soluble form-VEGF-B186 being more efficient.Citation13 VEGF-B167, with a better tissue specificity, might be more likely to fulfill the tissue-specific demand of FA uptake to cooperate with the oxidative capacity of specific tissue.Citation59 In summary, the VEGF-B creates a metabolic cross-talk between the ECs and the tissue cells, hence guaranteeing the energy accommodation and simultaneously tackling intracellular lipids accumulation and lipotoxicity.Citation60 However, the passively lipids transportation in obesity states can progressed into the insulin resistance and the subsequent T2DM.
Targeted VEGF-B therapy for T2DM
Due to its important roles in mediating lipid transport and metabolism, VEGF-B has been proposed as a novel therapeutic molecule for T2DM remedy via existing methods including gene deletion, gene slicing and the neutralizing monoclonal antibodies (mAbs). Both the construction of genetically engineered VEGF-B−/− modelCitation13,Citation61 and VEGF-B neutralizing bodies treatmentCitation13 ameliorated the diabetic phenotype, although the obesity phenotype remained. Many insights have been provided, despite in the animal models, providing the likelihood of developing the therapy tactics for the patients. In the VEGF-B−/− models,Citation13 the first phenotypic alteration observed is the declined levels of FATP3/4, resulting in the degressive lipids uptake, thereupon smaller and less abundant intracellular lipid droplets in heart, muscle, liver and BAT. Then, the accompanied promoted insulin sensitivity, glucose transporter-4 distribution on the membrane, glucose uptake; decreased plasma glucose and ameliorating glucose tolerance were observed, accordingly maintaining the euglycemia. These led to a compensatory amplification in carbohydrate utilized for energy production, indicating a metabolic transformation from FAs oxidation to glucose burning. Then, the unconsumed FAs were transferred to WAT, causing a growing body weight. Maybe the FAs uptake into the WATs is a process utilizing molecules other than VEGF-B?Citation59 Nonetheless, various VEGF-B deficient mouse strains exhibited inconsistent phenotypes in baseline conditions,Citation46 and the phenotypes described above are the comprehensive analysis of several studies.Citation13,Citation61 For the impact of VEGF-B deletion in HFD mice and in diabetic db/db mice, a gene dosage effect in males was observed since the VEGF-B+/− mice showed slightly elevated blood glucose levels compared with VEGF-B−/− mice.Citation61 Also, the VEGF-B−/− mice showed lower plasma triglycerides (TGs) and LDL/VLDL-bound cholesterol ratio, and higher levels of HDL-bound cholesterol, suggesting a lower risk for cardiovascular diseases (CVD).Citation61 Moreover, therapeutic inhibition of VEGF-B preserved islet functionality and insulin production by protecting islet architecture and guarding against β-cell apoptosis, possibly via the blunted lipotoxicity.Citation61 Antibody-mediated pharmacological controlling of VEGF-B phenocopied most of outcomes aforementioned. The neutralizing VEGF-B antibody (2H10) blocked the VEGF-B to binding its receptors, bringing about a long-lasting neutralization effect.Citation2
Despite these progresses, cautions should be taken when further translating to humans. Many T2DM patients also suffer from myocardial ischemia, coronary artery disease and diabetic neuropathy, thus would potentially benefit from VEGF-B coronary arteriogenic, neuron-protective and neurogenic effects.Citation46 Since the VEGF-B levels did not differ between T2DM patients and normal controls,Citation62 abrogating these effects may be detrimental. The tumor angiogenesis inhibiting effects also deserve cogitation.Citation46 Maybe the tissue specific deletion or the shRNA and the targeting delivery of anti-VEGF-B-body represent the possible curative orientation. A pinpointed understanding of the tissue/cell-specific expression pattern of VEGF-B and VEGFR-1 is necessary, and reporter-gene expressing models may help.Citation63
RobciucCitation64 et al. implied the promise of VEGF-B transgenic or VEGF-B protein delivery to ameliorate insulin sensitivity (to improve insulin sensitivity or to ameliorate insulin resistance), diminishes obesity, and alleviate metabolic syndrome, by displacing VEGF-A from VEGFR-1 to activate VEGFR-2 and increasing adipose tissue vascularity, thereupon providing a therapeutic tactics for counteracting obesity. Similar findings were reported in another report.Citation59 The contradiction aroused that how to settle the discrepancies between the two studies.Citation13,Citation64 We compared, in order to make them seem harmonious. (i) The insulin resistance in the two studies was derived from the insufficient vascularization and consecutive hypoxia and the ectopic fat accumulation, thus the VEGF-B linked downstream angiogenic event and the suppression of the VEGF-B medicated lipids trafficking could help, respectively. (ii) The VEGF-B gain-of-function may be applicable to the early period when the body owns the functional islets and normal insulin levels, mainly act on the adipose tissue; however, the VEGF-B loss-of-therapy is probably suitable for the terminal stages, targeting for the non-adipose tissue. (iii) Were the amelioration of the gain-of-therapy can be enlarged to the muscular tissues, such as muscle and liver, its suitability could be enlarged. If so, the possible side effect of the VEGF-B loss-of-function in the diabetes patient might be evaded and conversely can exert both anti-diabetic and neuron-protecting, cancer-inhibiting effects. (iv) Were restricted, along the deterioration of insulin resistance, the therapeutic regimen develops from the single gain-of-function to the combination of the gain/loss-of-function. Therefore, to suppress the VEGF-B in the muscular tissue and to enhance the role of VEGF-B in the adipose tissue simultaneously and to avoid the potential side-effect, a tissue-specific delivery of VEGF-B is required, which may be achieved by the newly designed tPep-VEGF-B targeting the adipose tissue.Citation65
Cardiovascular diseases, to achieve the tissue specific effect
Mickle studies have implicated the role of VEGF-B in cardiac development, indicating its potential therapeutic application in treating heart diseases. The heart owns the highest mRNA level of VEGF-BCitation13,Citation14 and the developing heart (pre- and postnatal) exhibited the primary VEGF related factor expression.Citation12,Citation66 During embryonic (E12.5-17.5) and early postnatal (P3) development, the most prominent cardiac VEGF-B expression changed from right to left ventricular wall,Citation67,Citation68 however the right ventricular wall after postnatal cardiac remodeling (P18) and in the adult, implying the coordination of its expression adaptation with the changes in cardiac energy requirements at various development stages.Citation63,Citation69 Gene knockout studies showed that VEGF-B does participate in coronary vasculature development and normal physiological responses to ischemia and vascular occlusion.Citation18 And, the HF patients showed the declined VEGF-B,Citation70 both in ischemic and dilated cardiomyopathy. Mice lacking VEGF-B displayed mild cardiac phenotypes, such as the slightly smaller heart and dysfunctional coronary vasculature in Bellomo VEGF-B−/− miceCitation67 and a prolonged PQ interval in Aase VEGF-B−/− mice.Citation71 Though minor phenotype differs, these inferred a protective role of VEGF-B in the normal or ischemic heart. Additional, individuals with diabetes are prone to suffer from CVD. Given the apparent role in the cardiac development and amelioration in the metabolic symptoms, VEGF-B therapy may show its appliance value in the HF patients.
Although several tactics are applied in the ischemic cardiomyopathy, such as acute coronary care, reperfusion of occluded coronary vessels and improvements in pharmacologic therapy, the mortality is still substantially ascending.Citation72 Genetic therapeutic vascular growth to induce the angiogenesis and arteriogenesis event may be an succedaneous approach for those with myocardial or peripheral ischemia who are unsuitable to conventional revascularization options.Citation73 simplified several VEGF-B gene transfer studies, providing us the insight of its protective role in myocardial ischemia and HF models.
Table 1. Summary of VEGF-B overexpression studies
Although VEGF-B is dispensable for maintaining normal cardiac capability under unstressed conditions,Citation17,Citation78 mounting evidence suggested distinct but complementary roles for VEGF-B in the maintenance of cardiac contractility and coronary perfusion.Citation70 Opposited to the pathological cardiac hypertrophy, the VEGF-B overexpression derived adaptive hypertrophy did not deteriorate into HF,Citation70 and this maybe also a paracrine event: the VEGFR-1 produced by the cardiomyocytesCitation79 however distributed on the ECs, establishing the ECs-Cardiomyocyte cross-talks. As for the metabolic altering, the strengthened MAPK and weakened AMPK signaling modulate cardiomyocytes to favor glucose oxidation and macromolecular biosynthesis.Citation70 The enhanced glucose utilization could avoid the ischemic myocardium and limit the myocardial ischemia/reperfusion injury, by augmenting the energy production in the energy-depleted myocardium.Citation80,Citation81 Coincidentally, such a metabolic shift was also occurred on the VEGF-B−/− mice,Citation13 and may be favoring the glucose oxidation is profitable for cardiac ability. The vascular growth also linked to the ECs metabolism.Citation82 Potentially, the exogenous VEGF-B expression can touch off the endogenous expression of co-expressed mitochondrial genes, given endogenous VEGF-B levels are highest in the heart.Citation8 The antiapoptotic impact was accounted to the antiapoptotic gene expression profile in cardiomyocytes and the metabolic change. To summarize, the effect of VEGF-B overexpression reflected in three aspects, cardiac growth promoting effect therefore displayed a compensatory hypertrophy; angiogenic effect and its related metabolic effects to modulate metabolism of ECsCitation82 and cardiomyocytes;Citation70 the protection of cardiomyocytes; from apoptosis; the manipulation of cardiac stem cell to protect against short- and long-term ischemia-reperfusion injury.Citation83 The combination of these yielded a prolonged beneficial influence on the heart, making the VEGF-B a promising candidate for the treatment of myocardial ischemia.
There are a few points worthy further investigations. First, a complementary relevancy existed between these effects: the cardiac hypertrophy could be the relevant consequence of the others. Vascularization is crucial for adaptive hypertrophy, as enlarged heart tissue must match with the concordant expansion of the coronary vasculature to maintain an adequate supply of oxygen and nutrients for the heart.Citation15,Citation84 The VEGF-B mediated lipids accumulation in the heart might also contributed, since inherited and acquired cardiomyopathies have the marked cardiac intracellular lipid accumulation.Citation85
Second, despite some similar phenotype changes were observed in two studies, discrepancy existed, even completely contradictory results. For example, in the two studies,Citation70,Citation77 the common myocardial-specific angiogenesis and arteriogenes activity were observed. Whereas, in rabbits and the pigs,Citation77 the aggrandizement of FATP4 expression and lipids and glycogen accumulation in the myocardium were observed; while there is no difference in cardiac or skeletal muscle FAs influx between the VEGF-B transgenic, gene-deleted and wild type rats.Citation70 It is important to note that FAs and TGs levels were reduced in the transgenic rat. The reason might be the FATP4 was actually a fatty Acyl-CoA synthase,Citation86 which meant the FATP4 directs the FAs to synthetic pathways rather than oxidation.
Third, the diverse mechanisms may lead in the same myocardial hypertrophy phenotype. For example, the transgenic mice developed an invalid cardiac hypertrophy due to an enlarged size of the cardiomyocytes but lacked an arteriogenic phenotype, failed to compromise heart ability;Citation15 however, the cardiac hypertrophy observed in the transgenic or AAV-VEGF-B overexpression rats could be attributed to the coronary tree expanding and reprogram of the cardiomyocyte energy substrate utilization from FAs oxidation towards glucose oxidation.Citation70 In turn, the alike manifold lipid and glycogen accumulation in the myocardium caused the metabolic changes,Citation77 cardiac hypertrophy,Citation70 mitochondrial lipotoxicity and the consecutive mouse death,Citation15 respectively in three distinct studies.
Furthermore, just noticing the angiogenic role of VEGF-B in vivo, an apparent disagreement exists ranging from no angiogenic ability at all in several tissues after adenovirus gene delivery;Citation87 ability to potentiate rather than bringing about angiogenesis when transduced into the endothelial barrier;Citation88 a restricted revascularization in the ischemic myocardium.Citation18,Citation77 Several factors had been proposed to participate, such as the diversity of genetic background, VEGF-B isoforms or means for VEGF-B overproduction (recombinant proteins, naked plasmid DNA or adenoviral vectors).Citation89 In this respect, adenoviruses and AAV vectors administration represented the most efficient vectors to transfer genes into the adult myocardium and ensured a prolonged effect.Citation90 The AAV vectors had acquired increasing popularity due to its ability to transduce postmitotic cells, such as cardiomyocytes, at high efficiency and to drive lasting periods of gene expression with noticeable inflammation.Citation91 Other vector systems, such as the first generation adenoviruses, could activate innate immune responses.Citation92
Regarding the VEGF-B as an endogenous protective and repair-promoting cardiac proteins, its overexpression can be efficacious to prevent cardiac damage and enhance tissue repair.Citation93 The other members, VEGF-ACitation94 and PlGFCitation95 also showed protective ability. Howbeit, the risk of adverse effects, consisting of bleeding, leakage, hypotension, malignancy, had limited clinically systemic administration of VEGF-A for the revascularization of ischemic tissues;Citation96 and the Ad-PlGF induced myocardial angiogenesis and cardiac hypertrophy was abolished by the nitric oxide synthase inhibitor L-arginine Methyl Ester (L-NAME).Citation97 The L-NAME did not cancel VEGF-B activity, emphasizing the uniqueness of VEGF-B. To explain these different effects, Ad-VEGF-B186 and Ad-PlGF might signal through different receptor binding sites and/or structural variants, or alternatively, acted by recruiting distinct co-receptors to the signaling complex,Citation13 thereby inducing various downstream events,Citation77 which was verified since VEGF-B did not rescue development in PlGF deficiency mice.Citation95 In comparison, the VEGF-B has marked superiority over its family members. First, VEGF-B showed high selectivity to stimulate angiogenesis, especially in the ischemic myocardium,Citation18 being expected to stimulate angiogenic without causing adverse effects. The upregulation of VEGF-B levels in the ischemic heart but not in the ischemic muscle might partly account for its specificity. Another possible mechanism might be the distinct ECs differentiation in the isolated tissue and organs.Citation98 Then, VEGF-B provided a more balanced vascularization, embracing microvessel maturation, arteriogenesis besides mere angiogenesis. Nevertheless, the rAAV-VEGF-A barely led to vessel formation but failed to enhance collateralization and perfusion, unless platelet-derived growth factor-B was co-transfected.Citation99 Moreover, high amounts of VEGF-B were well tolerated, predicating the much wider therapeutic window than VEGF-A or VEGF-C.Citation70 This uniqueness of VEGF-B warrant further cogitation of the therapeutic potential of VEGF-B for promoting functional recovery of myocardial ischemia.
Neurodegenerative disorders, potent protection without angiogenic by- effect
VEGF-B also manifested its safeguarding role in the neurodegenerative disorders. Indeed, the up-regulated transcriptional activation of VEGF-B in response to midbrain neurodegenerative challenges was observed in Parkinson's Disease (PD), and VEGF-B produced by astrocytes and motor neurons exerted a neuroprotective affect.Citation22,Citation100 Preclinical studies in PDCitation101 and amyotrophic lateral sclerosis modelCitation22 had shown promising results, despite the lack of clinical studies. VEGF-B treatment rescued brain neurons from apoptosis in stroke mouseCitation21 and protected cultured primary motor neurons against degeneration,Citation22 with little retinal neovascularization effect. The co-expression of VEGF-B with FAs oxidation relative mitochondrial genes was observed in rat midbrain, suggesting the mitochondria as the target site.Citation102 Moreover, VEGF-B possessed the considerably robust survival ability,Citation101,Citation102 as a single VEGF-B186 protein treatment at a dose of 3 mg per rat partially protected dopaminergic fibers in the striatum and rescued the dopaminergic neurons in the caudal sub-region of the substantianigra.Citation101
The neurobiological activity of VEGF-A consists of neuroprotection, neurogenesis, and angiogenesis.Citation103 Whereas, VEGF-B lacks the undesired adverse angiogenic vitality,Citation100 consequently it could be regarded as a trophic divisor to reduce effects of neurodegeneration. And, the VEGF-B had no visible neurorestoration effect.Citation102 Given the tempting neuroprotective activity combined with negligible angiogenic/permeability activity,Citation21,Citation22 strategies such as adding exogenous VEGF-B or up-regulating the endogenous VEGF-B levels to strengthen this natural protective response may have the potential to be a disease modifying therapy for PD.Citation100
Cancer, dual effects on cancer metastasis and growth
The correlation of VEGF-B with cancer remains unclear. Considering the cancer tissues had the higher VEGF-B level,Citation104 The VEGF-B might promote the cancer progression, especially in advanced cancers. However, the VEGF-B also retarded tumor growth in the RIP1-Tag2 mouse of pancreatic neuroendocrine tumorigenesis,Citation6 and the reduced blood perfusion in VEGF-B-T241 tumors might explain, at least in part, the anti-tumor growth effect.Citation105 The antigrowth effect might also be accounted for the approximate 15% heavier weight displayed in the VEGF-B−/− mice.Citation6,Citation13 The VEGFR-1 may explained the antigrowth and antiangiogenic effect, since it is served as the decoy receptor of VEGF-A.Citation106 The VEGFR-1 had a negative role in developmental vascularization, and VEGFR-1−/− embryos died early due to VEGF-A dependent vessels overgrowth and disorganization.Citation107 Moreover, the VEGF-B advanced tumor invasiveness both in HCC patientsCitation108 and mouse tumors,Citation105 via remodeling of the tumor microvasculature, leading to leaky vascular networks that are highly permissive for invasion.Citation105 So, VEGF-B might have paradoxical roles in cancer initiation and further progression, inhibiting growth and promoting metastasis, which insinuated the uncoupling of the metastasis and primary tumor growth.Citation105
Overall, the tumor retains the higher VEGF-B levels, associated with high rates of distant failure and poor overall survival,Citation109 and it was responsible to expect that lowering the VEGF-B may show anti-cancer effect. Actually, the recent reported metformin made it.Citation110 To let down it, several methods have been put forward. The gene deletion therapy can provide the prolonged effect though the tissue specific deletion barriers and the existing physical function of VEGF-B astricted its application. Similar therapeutic outcomes can be achieved by administrating monoclonal antibodies. Given its potential role for in vivo function characterization and the identification for the new therapeutic strategies, more therapeutic antibodies against human VEGF-B, and small molecule tyrosine kinase inhibitors are deserved to be raised.
Concluding remarks and future prospects
VEGF-B is inert under physiological conditions while showing a potent and safe therapeutic potential in treating metabolic dysregulations, correlative with its widely distribution and multifaceted features. These seeming contradictory features enable the VEGF-B the valuable therapeutic significance in clinical at an attractive safety profile, even the feasibility of developing into the drugs.
The studies of recombinant VEGF-B protein had been limited, due to the burdens in the purification and the lacking of VEGFR-1 mediated responses that might form the basis of a simple cell-based assay system.Citation14 The exist for VEGF-B purification are largely based on affinity chromatography.Citation65,Citation111 Comparing with the molecular pharmacological interventions, gene therapy may provide a long-lasting therapeutic effect.Citation72 In terms of the mAbs, more studies are warranted to interpret the intrinsic molecular basis and to design new molecules with optimized pharmacokinetics/pharmacodynamics, given a series of variables such as potency, half-life, binding stability, bioavailability, and dosing regimen of the existing VEGF-A blockers reflected in clinical efficacyCitation112 and the complex relationship between clinical studies.Citation113 And, considering the unsuspected feed-back loops and cross-talk between diversified signaling pathways, the efficacy of conventional molecule has been less than expected, there by the design of mAbs that targeting multiple pathways, especially the intracrine (intracellular and autocrine) signaling pathways, perhaps be an optional orientation, such as epidermal growth factor receptor-VEGF(R) pathway cross-talk in the cancer angiogenesis.Citation114
Nevertheless, it is just the multiple features that also limit its application, such as the side effect of the VEGF-B in diabetes. Among these models, diabetes represents the most promising, however others are limited to the rodent studies. More studies are under consideration to uncover the more precise role of VEGF-B under physiological conditions and its possible application in treating diseases. How does the context-dependent varied features switch occur? How it achieved the cardiac specific angiogenesis effect? Also, were the VEGF-B gain-of-therapy enlarged its ability to the muscular tissues, how to actualize the cell/tissue specific delivery of the VEGF-B or its antibodies to avert the possible by-effect, and the development of the long-acting analogs or some other VEGF mimetics are also worth exploring. The clarify of the cross-talk of the family members, if necessary be specific to the tissue, is also a huge task in the long time.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was funded in part by the China National Nature Science Foundation (81302685, 81430082), the Hi-Tech Research and Development Program of China-863 Program (2015AA020945), the Innovative Scientific Research Team Fund of Jiangsu Province, the Open Project of State Key Laboratory of Natural Medicines, No. SKLNMKF201709 and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
- Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21:154-65. doi:10.1016/j.ceb.2008.12.012. PMID:19230644
- Leonard P, Scotney PD, Jabeen T, Iyer S, Fabri LJ, Nash AD, Acharya KR. Crystal structure of vascular endothelial growth factor-B in complex with a neutralising antibody Fab fragment. J Mol Biol. 2008;384:1203-17. doi:10.1016/j.jmb.2008.09.076. PMID:18930733
- Stuttfeld E, Ballmer-Hofer K. Structure and function of VEGF receptors. IUBMB Life. 2009;61:915-22. doi:10.1002/iub.234. PMID:19658168
- Neufeld G, Kessler O, Herzog Y. The interaction of Neuropilin-1 and Neuropilin-2 with tyrosine-kinase receptors for VEGF. Adv Exp Med Biol. 2002;515:81-90. doi:10.1007/978-1-4615-0119-0_7. PMID:12613545
- Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem 1994;269:25646-54. PMID:7929268
- Albrecht I, Kopfstein L, Strittmatter K, Schomber T, Falkevall A, Hagberg CE, Lorentz P, Jeltsch M, Alitalo K, Eriksson U, et al. Suppressive effects of vascular endothelial growth factor-B on tumor growth in a mouse model of pancreatic neuroendocrine tumorigenesis. PLoS One. 2010;5:e14109. doi:10.1371/journal.pone.0014109. PMID:21124841
- During MJ, Liu X, Huang W, Magee D, Slater A, McMurphy T, Wang C, Cao L. Adipose VEGF Links the White-to-Brown Fat Switch With Environmental, Genetic, and Pharmacological Stimuli in Male Mice. Endocrinology. 2015;156:2059-73. doi:10.1210/en.2014-1905. PMID:25763639
- Olofsson B, Pajusola K, Kaipainen A, von Euler G, Joukov V, Saksela O, Orpana A, Pettersson RF, Alitalo K, Eriksson U. Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc Natl Acad Sci U S A 1996;93:2576-81. doi:10.1073/pnas.93.6.2576. PMID:8637916
- Li X, Kumar A, Zhang F, Lee C, Tang Z. Complicated life, complicated VEGF-B. Trends Mol Med. 2012;18:119-27. doi:10.1016/j.molmed.2011.11.006. PMID:22178229
- Scotney PD, MacKenzie A, Maccarone P, Fabri LJ, Scrofani SD, Gooley PR, Nash AD. Human vascular endothelial growth factor B: characterization of recombinant isoforms and generation of neutralizing monoclonal antibodies. Clin Exp Pharmacol Physiol. 2002;29:1024-9. doi:10.1046/j.1440-1681.2002.03769.x. PMID:12366396
- Olofsson B, Pajusola K, von Euler G, Chilov D, Alitalo K, Eriksson U. Genomic organization of the mouse and human genes for vascular endothelial growth factor B (VEGF-B) and characterization of a second splice isoform. J Biol Chem 1996;271:19310-7. doi:10.1074/jbc.271.32.19310. PMID:8702615
- Aase K, Lymboussaki A, Kaipainen A, Olofsson B, Alitalo K, Eriksson U. Localization of VEGF-B in the mouse embryo suggests a paracrine role of the growth factor in the developing vasculature. Dev Dyn. 1999;215:12-25. doi:10.1002/(SICI)1097-0177(199905)215:1<12::AID-DVDY3>3.0.CO;2-N. PMID:10340753
- Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, van Meeteren LA, Samen E, Lu L, Vanwildemeersch M, et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917-21. doi:10.1038/nature08945. PMID:20228789
- Nash AD, Baca M, Wright C, Scotney PD. The biology of vascular endothelial growth factor-B (VEGF-B). Pulm Pharmacol Ther. 2006;19:61-9. doi:10.1016/j.pupt.2005.02.007. PMID:16286239
- Karpanen T, Bry M, Ollila HM, Seppanen-Laakso T, Liimatta E, Leskinen H, Kivelä R, Helkamaa T, Merentie M, Jeltsch M, et al. Overexpression of vascular endothelial growth factor-B in mouse heart alters cardiac lipid metabolism and induces myocardial hypertrophy. Circ Res. 2008;103:1018-26. doi:10.1161/CIRCRESAHA.108.178459. PMID:18757827
- Rissanen TT, Markkanen JE, Gruchala M, Heikura T, Puranen A, Kettunen MI, Kholová I, Kauppinen RA, Achen MG, Stacker SA, et al. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res. 2003;92:1098-106. doi:10.1161/01.RES.0000073584.46059.E3. PMID:12714562
- Reichelt M, Shi S, Hayes M, Kay G, Batch J, Gole GA, Browning J. Vascular endothelial growth factor-B and retinal vascular development in the mouse. Clin Exp Ophthalmol. 2003;31:61-5. doi:10.1046/j.1442-9071.2003.00602.x. PMID:12580897
- Li X, Tjwa M, Van Hove I, Enholm B, Neven E, Paavonen K, Jeltsch M, Juan TD, Sievers RE, Chorianopoulos E, et al. Reevaluation of the role of VEGF-B suggests a restricted role in the revascularization of the ischemic myocardium. Arterioscler Thromb Vasc Biol. 2008;28:1614-20. doi:10.1161/ATVBAHA.107.158725. PMID:18511699
- Zhong XF, Huang H, Shen JK, Zacchigna S, Zentilin L, Giacca M, Vinores SA. Vascular endothelial growth factor-B gene transfer exacerbates retinal and choroidal neovascularization and vasopermeability without promoting inflammation. Mol Vis. 2011;17:492-507. PMID:21364963
- Claesson-Welsh L. VEGF-B Taken to Our Hearts Specific Effect of VEGF-B in Myocardial Ischemia. Arterioscl Throm Vas. 2008;28:1575-6. doi:10.1161/ATVBAHA.108.170878.
- Li Y, Zhang F, Nagai N, Tang Z, Zhang S, Scotney P, Lennartsson J, Zhu C, Qu Y, Fang C, et al. VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J Clin Invest. 2008;118:913-23. PMID:18259607
- Poesen K, Lambrechts D, Van Damme P, Dhondt J, Bender F, Frank N, Bogaert E, Claes B, Heylen L, Verheyen A, et al. Novel role for vascular endothelial growth factor (VEGF) receptor-1 and its ligand VEGF-B in motor neuron degeneration. J Neurosci. 2008;28:10451-9. doi:10.1523/JNEUROSCI.1092-08.2008. PMID:18923022
- Eichmann A, Makinen T, Alitalo K. Neural guidance molecules regulate vascular remodeling and vessel navigation. Genes Dev. 2005;19:1013-21. doi:10.1101/gad.1305405. PMID:15879551
- Li X, Lee C, Tang Z, Zhang F, Arjunan P, Li Y, Hou X, Kumar A, Dong L. VEGF-B: a survival, or an angiogenic factor? Cell Adh Migr. 2009;3:322-7. doi:10.4161/cam.3.4.9459. PMID:19684473
- Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148:1160-71. doi:10.1016/j.cell.2012.02.010. PMID:22424227
- A DR. The triumvirate: β-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1998;37:667-87.
- Bergman RN. Minimal model: perspective from 2005. Horm Res. 2005;64 Suppl 3:8-15. PMID:16439839
- Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011;54:2506-14. doi:10.1007/s00125-011-2204-7. PMID:21656330
- Phielix E, Meex R, Ouwens DM, Sparks L, Hoeks J, Schaart G, Moonen-Kornips E, Hesselink MK, Schrauwen P. High oxidative capacity due to chronic exercise training attenuates lipid-induced insulin resistance. Diabetes. 2012;61:2472-8. doi:10.2337/db11-1832. PMID:22787138
- Deacon CF, Carr RD, Holst JJ. DPP-4 inhibitor therapy: new directions in the treatment of type 2 diabetes. Front Biosci. 2008;13:1780-94. doi:10.2741/2799. PMID:17981667
- Group EDP. A desktop guide to Type 2 diabetes mellitus. European Diabetes Policy Group 1999. Diabet Med. 1999;16:716-30. PMID:10510947
- Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963;1:785-9. doi:10.1016/S0140-6736(63)91500-9. PMID:13990765
- Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375:2267-77. doi:10.1016/S0140-6736(10)60408-4. PMID:20609972
- Turinsky J, O'Sullivan DM, Bayly BP. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem. 1990;265:16880-5. PMID:2211599
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277: 50230-6. doi:10.1074/jbc.M200958200. PMID:12006582
- Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001;50:2563-71. doi:10.2337/diabetes.50.11.2563. PMID:11679435
- Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 1999;42:113-6. doi:10.1007/s001250051123. PMID:10027589
- Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852-71. doi:10.1016/j.cell.2012.02.017. PMID:22385956
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793-801. doi:10.1172/JCI29069. PMID:16823477
- Kaneto H, Nakatani Y, Kawamori D, Miyatsuka T, Matsuoka TA, Matsuhisa M, Yamasaki Y. Role of oxidative stress, endoplasmic reticulum stress, and c-Jun N-terminal kinase in pancreatic beta-cell dysfunction and insulin resistance. Int J Biochem Cell Biol. 2006;38:782-93. doi:10.1016/j.biocel.2006.01.004. PMID:16607699
- Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621-37. doi:10.1172/JCI31021. PMID:17717599
- Stefanovski D, Richey JM, Woolcott O, Lottati M, Zheng D, Harrison LN, Ionut V, Kim SP, Hsu I, Bergman RN. Consistency of the disposition index in the face of diet induced insulin resistance: potential role of FFA. PLoS One. 2011;6:e18134. doi:10.1371/journal.pone.0018134. PMID:21479217
- Hoppa MB, Collins S, Ramracheya R, Hodson L, Amisten S, Zhang Q, Johnson P, Ashcroft FM, Rorsman P. Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca(2+) channels from secretory granules. Cell Metab. 2009;10:455-65. doi:10.1016/j.cmet.2009.09.011. PMID:19945403
- Tushuizen ME, Bunck MC, Pouwels PJ, Bontemps S, van Waesberghe JH, Schindhelm RK, Mari A, Heine RJ, Diamant M. Pancreatic fat content and β-cell function in men with and without type 2 diabetes. Diabetes care. 2007;30:2916-21. doi:10.2337/dc07-0326. PMID:17666465
- Collins SC, Hoppa MB, Walker JN, Amisten S, Abdulkader F, Bengtsson M, Fearnside J, Ramracheya R, Toye AA, Zhang Q, et al. Progression of diet-induced diabetes in C57BL6J mice involves functional dissociation of Ca2(+) channels from secretory vesicles. Diabetes. 2010;59:1192-201. doi:10.2337/db09-0791. PMID:20150285
- Carmeliet P, Wong BW, De Bock K. Treating diabetes by blocking a vascular growth factor. Cell Metab. 2012;16:553-5. doi:10.1016/j.cmet.2012.10.015. PMID:23140637
- Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104:12587-94. doi:10.1073/pnas.0705408104. PMID:17640906
- Hazlehurst JM, Woods C, Marjot T, Cobbold JF, Tomlinson JW. Non-alcoholic fatty liver disease and diabetes. Metabolism. 2016;65:1096-108. doi:10.1016/j.metabol.2016.01.001. PMID:26856933
- Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26:244-51. doi:10.1016/j.tips.2005.03.003. PMID:15860371
- Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med. 1994;331:1188-93. doi:10.1056/NEJM199411033311803. PMID:7935656
- Yang X, Smith U. Adipose tissue distribution and risk of metabolic disease: Does thiazolidinedione-induced adipose tissue redistribution provide a clue to the answer? Diabetologia 2007;50(6):1127-39. doi: 10.1007/s00125-007-0640-1. PMID: 17393135
- Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993-9. doi:10.1016/j.cell.2005.11.026. PMID:16360030
- Mehrotra D, Wu J, Papangeli I, Chun HJ. Endothelium as a gatekeeper of fatty acid transport. Trends Endocrin Met. 2014;25:99-106. doi:10.1016/j.tem.2013.11.001.
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457-71. doi:10.1056/NEJMoa072761. PMID:17517853
- Miller CP. SERMs: evolutionary chemistry, revolutionary biology. Curr Pharm Des. 2002;8:2089-111. doi:10.2174/1381612023393404. PMID:12171520
- Hegarty BD, Furler SM, Oakes ND, Kraegen EW, Cooney GJ. Peroxisome proliferator-activated receptor (PPAR) activation induces tissue-specific effects on fatty acid uptake and metabolism in vivo—a study using the novel PPARα/γ agonist tesaglitazar. Endocrinology. 2004;145:3158-64. doi:10.1210/en.2004-0260. PMID:15059948
- Hagberg C, Mehlem A, Falkevall A, Muhl L, Eriksson U. Endothelial fatty acid transport: role of vascular endothelial growth factor B. Physiology (Bethesda). 2013;28:125-34. doi:10.1152/physiol.00042.2012. PMID:23455771
- Mehlem A, Palombo I, Wang X, Hagberg CE, Eriksson U, Falkevall A. PGC-1alpha Coordinates Mitochondrial Respiratory Capacity and Muscular Fatty Acid Uptake via Regulation of VEGF-B. Diabetes. 2016;65:861-73. doi:10.2337/db15-1231. PMID:26822083
- Li X. VEGF-B: a thing of beauty. Cell Res. 2010;20:741-4. doi:10.1038/cr.2010.77. PMID:20531376
- Muoio DM, Koves TR. Lipid-induced metabolic dysfunction in skeletal muscle. Novartis Found Symp 2007; 286:24-38; discussion-46, 162-3, 96-203. doi: 10.1002/9780470985571.ch4. PMID:18269172
- Hagberg CE, Mehlem A, Falkevall A, Muhl L, Fam BC, Ortsater H, Scotney P, Nyqvist D, Samén E, Lu L, et al. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature. 2012;490:426-30. doi:10.1038/nature11464. PMID:23023133
- Sun CY, Lee CC, Hsieh MF, Chen CH, Chou KM. Clinical association of circulating VEGF-B levels with hyperlipidemia and target organ damage in type 2 diabetic patients. J Biol Regul Homeost Agents. 2014;28:225-36. PMID:25001655
- Muhl L, Moessinger C, Adzemovic MZ, Dijkstra MH, Nilsson I, Zeitelhofer M, Hagberg CE, Huusko J, Falkevall A, Ylä-Herttuala S, et al. Expression of vascular endothelial growth factor (VEGF)-B and its receptor (VEGFR1) in murine heart, lung and kidney. Cell Tissue Res. 2016;365:51-63. doi:10.1007/s00441-016-2377-y. PMID:26928042
- Robciuc MR, Kivela R, Williams IM, de Boer JF, van Dijk TH, Elamaa H, Tigistu-Sahle F, Molotkov D, Leppänen VM, Käkelä R, et al. VEGFB/VEGFR1-Induced Expansion of Adipose Vasculature Counteracts Obesity and Related Metabolic Complications. Cell Metab. 2016;23:712-24. doi:10.1016/j.cmet.2016.03.004. PMID:27076080
- Zhang Y, Tong Y, Gao M, Luo C, Song X, Gao X, Yao W. Expression, purification and characterization of a vascular endothelial growth factor fusion protein. Biotechnol Lett. 2016;38:1115-20. doi:10.1007/s10529-016-2081-8. PMID:26976430
- Lagercrantz J, Larsson C, Grimmond S, Fredriksson M, Weber G, Piehl F. Expression of the VEGF-related factor gene in pre- and postnatal mouse. Biochem Biophys Res Commun. 1996;220:147-52. doi:10.1006/bbrc.1996.0372. PMID:8602835
- Bellomo D, Headrick JP, Silins GU, Paterson CA, Thomas PS, Gartside M, Mould A, Cahill MM, Tonks ID, Grimmond SM, et al. Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature, and impaired recovery from cardiac ischemia. Circ Res. 2000;86:e29−e35. doi:10.1161/01.RES.86.2.e29. PMID:10666423
- Lagercrantz J, Farnebo F, Larsson C, Tvrdik T, Weber G, Piehl F. A comparative study of the expression patterns for vegf, vegf-b/vrf and vegf-c in the developing and adult mouse. Biochim Biophys Acta 1998;1398:157-63. doi:10.1016/S0167-4781(98)00040-2. PMID:9689915
- Hew KW, Keller KA. Postnatal anatomical and functional development of the heart: a species comparison. Birth Defects Res B Dev Reprod Toxicol. 2003;68:309-20. doi:10.1002/bdrb.10034. PMID:14666994
- Kivela R, Bry M, Robciuc MR, Rasanen M, Taavitsainen M, Silvola JM, Saraste A, Hulmi JJ, Anisimov A, Mäyränpää MI, et al. VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol Med. 2014;6:307-21. PMID:24448490
- Aase K, von Euler G, Li X, Ponten A, Thoren P, Cao R, Cao Y, Olofsson B, Gebre-Medhin S, Pekny M, et al. Vascular endothelial growth factor-B-deficient mice display an atrial conduction defect. Circulation. 2001;104:358-64. doi:10.1161/01.CIR.104.3.358. PMID:11457758
- Kupatt C, Hinkel R. VEGF-B: a more balanced approach toward cardiac neovascularization? EMBO Mol Med. 2014;6:297-8. doi:10.1002/emmm.201303730. PMID:24521742
- Yla-Herttuala S, Alitalo K. Gene transfer as a tool to induce therapeutic vascular growth. Nat Med. 2003;9:694-701. doi:10.1038/nm0603-694. PMID:12778168
- Serpi R, Tolonen AM, Huusko J, Rysa J, Tenhunen O, Yla-Herttuala S, Ruskoaho H. Vascular endothelial growth factor-B gene transfer prevents angiotensin II-induced diastolic dysfunction via proliferation and capillary dilatation in rats. Cardiovasc Res. 2011;89:204-13. doi:10.1093/cvr/cvq267. PMID:20733007
- Bry M, Kivela R, Holopainen T, Anisimov A, Tammela T, Soronen J, Silvola J, Saraste A, Jeltsch M, Korpisalo P, et al. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725-33. doi:10.1161/CIRCULATIONAHA.110.957332. PMID:20937974
- Zentilin L, Puligadda U, Lionetti V, Zacchigna S, Collesi C, Pattarini L, Ruozi G, Camporesi S, Sinagra G, Pepe M, et al. Cardiomyocyte VEGFR-1 activation by VEGF-B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB J. 2010;24:1467-78. doi:10.1096/fj.09-143180. PMID:20019242
- Lähteenvuo JE, Lähteenvuo MT, Kivelä A, Rosenlew C, Falkevall A, Klar J, Heikura T, Rissanen TT, Vähäkangas E, Korpisalo P, et al. Vascular endothelial growth factor-B induces myocardium-specific angiogenesis and arteriogenesis via vascular endothelial growth factor receptor-1–and neuropilin receptor-1–dependent mechanisms. Circulation. 2009;119:845-56. doi:10.1161/CIRCULATIONAHA.108.816454. PMID:19188502
- Dijkstra MH, Pirinen E, Huusko J, Kivela R, Schenkwein D, Alitalo K, Ylä-Herttuala S. Lack of cardiac and high-fat diet induced metabolic phenotypes in two independent strains of Vegf-b knockout mice. Sci Rep. 2014;4:6238. doi:10.1038/srep06238. PMID:25168313
- Huusko J, Merentie M, Dijkstra MH, Ryhanen MM, Karvinen H, Rissanen TT, Vanwildemeersch M, Hedman M, Lipponen J, Heinonen SE, et al. The effects of VEGF-R1 and VEGF-R2 ligands on angiogenic responses and left ventricular function in mice. Cardiovasc Res. 2010;86:122-30. doi:10.1093/cvr/cvp382. PMID:19955220
- Vogt AM, Elsässer A, Nef H, Bode C, Kübler W, Schaper J. Increased glycolysis as protective adaptation of energy depleted, degenerating human hibernating myocardium. Mol Cell Biochem. 2003;242:101-7. doi:10.1023/A:1021141812947. PMID:12619871
- McLean BA, Kienesberger PC, Wang W, Masson G, Zhabyeyev P, Dyck JR, Oudit GY. Enhanced recovery from ischemia-reperfusion injury in PI3Kalpha dominant negative hearts: investigating the role of alternate PI3K isoforms, increased glucose oxidation and MAPK signaling. J Mol Cell Cardiol. 2013;54:9-18. doi:10.1016/j.yjmcc.2012.10.015. PMID:23142539
- Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231-44. doi:10.1161/CIRCRESAHA.116.302855. PMID:25814684
- Li GH, Luo B, Lv YX, Zheng F, Wang L, Wei MX, Li XY, Zhang L, Wang JN, Chen SY, et al. Dual effects of VEGF-B on activating cardiomyocytes and cardiac stem cells to protect the heart against short- and long-term ischemia-reperfusion injury. J Transl Med. 2016;14:116. doi:10.1186/s12967-016-0847-3. PMID:27146579
- Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108-18. doi:10.1172/JCI24682. PMID:16075055
- Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813-22. doi:10.1172/JCI10947. PMID:11285300
- Digel M, Staffer S, Ehehalt F, Stremmel W, Ehehalt R, Füllekrug J. FATP4 contributes as an enzyme to the basal and insulin-mediated fatty acid uptake of Cx2C12 muscle cells. Am J Physiol-Endoc M. 2011;301:E785-E96.
- Bhardwaj S, Roy H, Gruchala M, Viita H, Kholova I, Kokina I, Achen MG, Stacker SA, Hedman M, Alitalo K, et al. Angiogenic responses of vascular endothelial growth factors in periadventitial tissue. Hum Gene Ther. 2003;14:1451-62. doi:10.1089/104303403769211664. PMID:14577925
- Mould AW, Greco SA, Cahill MM, Tonks ID, Bellomo D, Patterson C, Zournazi A, Nash A, Scotney P, Hayward NK, et al. Transgenic overexpression of vascular endothelial growth factor-B isoforms by endothelial cells potentiates postnatal vessel growth in vivo and in vitro. Circ Res. 2005;97:e60−70. doi:10.1161/01.RES.0000182631.33638.77. PMID:16109918
- Goren HJ, Kulkarni RN, Kahn CR. Glucose homeostasis and tissue transcript content of insulin signaling intermediates in four inbred strains of mice: C57BL/6, C57BLKS/6, DBA/2, and 129X1. Endocrinology. 2004;145:3307-23. doi:10.1210/en.2003-1400. PMID:15044376
- Rissanen TT, Yla-Herttuala S. Current status of cardiovascular gene therapy. Mol Ther. 2007;15:1233-47. doi:10.1038/sj.mt.6300175. PMID:17505481
- Favre D, Provost N, Blouin V, Blancho G, Cherel Y, Salvetti A, Moullier P. Immediate and long-term safety of recombinant adeno-associated virus injection into the nonhuman primate muscle. Mol Ther. 2001;4:559-66. doi:10.1006/mthe.2001.0494. PMID:11735340
- Liu Q, Muruve DA. Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 2003;10:935-40. doi:10.1038/sj.gt.3302036. PMID:12756413
- Doroudgar S, Glembotski CC. The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol Med. 2011;17:207-14. doi:10.1016/j.molmed.2010.12.003. PMID:21277256
- Ferrarini M, Arsic N, Recchia FA, Zentilin L, Zacchigna S, Xu X, Linke A, Giacca M, Hintze TH. Adeno-associated virus-mediated transduction of VEGF165 improves cardiac tissue viability and functional recovery after permanent coronary occlusion in conscious dogs. Circ Res. 2006;98:954-61. doi:10.1161/01.RES.0000217342.83731.89. PMID:16543500
- Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7:575-83. doi:10.1038/87904. PMID:11329059
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932-6. doi:10.1038/nature04478. PMID:16355210
- Jaba IM, Zhuang ZW, Li N, Jiang Y, Martin KA, Sinusas AJ, Papademetris X, Simons M, Sessa WC, Young LH, et al. NO triggers RGS4 degradation to coordinate angiogenesis and cardiomyocyte growth. J Clin Invest. 2013;123:1718-31. doi:10.1172/JCI65112. PMID:23454748
- Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158-73. doi:10.1161/01.RES.0000255691.76142.4a. PMID:17272818
- Kupatt C, Hinkel R, Pfosser A, El-Aouni C, Wuchrer A, Fritz A, Globisch F, Thormann M, Horstkotte J, Lebherz C, et al. Cotransfection of vascular endothelial growth factor-A and platelet-derived growth factor-B via recombinant adeno-associated virus resolves chronic ischemic malperfusion role of vessel maturation. J Am Coll Cardiol. 2010;56:414-22. doi:10.1016/j.jacc.2010.03.050. PMID:20650363
- Falk T, Zhang S, Sherman SJ. Vascular endothelial growth factor B (VEGF-B) is up-regulated and exogenous VEGF-B is neuroprotective in a culture model of Parkinson's disease. Mol Neurodegener. 2009;4:49. doi:10.1186/1750-1326-4-49. PMID:20003314
- Falk T, Yue X, Zhang S, McCourt AD, Yee BJ, Gonzalez RT, Sherman SJ. Vascular endothelial growth factor-B is neuroprotective in an in vivo rat model of Parkinson's disease. Neurosci Lett. 2011;496:43-7. doi:10.1016/j.neulet.2011.03.088. PMID:21507340
- Yue X, Hariri DJ, Caballero B, Zhang S, Bartlett MJ, Kaut O, Mount DW, Wüllner U, Sherman SJ, Falk T. Comparative study of the neurotrophic effects elicited by VEGF-B and GDNF in preclinical in vivo models of Parkinson's disease. Neuroscience. 2014;258:385-400. doi:10.1016/j.neuroscience.2013.11.038. PMID:24291725
- Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111:1843-51. doi:10.1172/JCI200317977. PMID:12813020
- Eggert A, Ikegaki N, Kwiatkowski J, Zhao HQ, Brodeur GM, Himelstein BP. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res. 2000;6:1900-8. PMID:10815914
- Yang X, Zhang Y, Hosaka K, Andersson P, Wang J, Tholander F, Cao Z, Morikawa H, Tegnér J, Yang Y, et al. VEGF-B promotes cancer metastasis through a VEGF-A-independent mechanism and serves as a marker of poor prognosis for cancer patients. Proc Natl Acad Sci U S A. 2015;112:E2900−9. doi:10.1073/pnas.1503500112. PMID:25991856
- Shibuya M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis. 2006;9:225-30; discussion 31. doi:10.1007/s10456-006-9055-8. PMID:17109193
- Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995;376:66-70. doi:10.1038/376066a0. PMID:7596436
- Kanda M, Nomoto S, Nishikawa Y, Sugimoto H, Kanazumi N, Takeda S, Nakao A. Correlations of the expression of vascular endothelial growth factor B and its isoforms in hepatocellular carcinoma with clinico-pathological parameters. J Surg Oncol. 2008;98:190-6. doi:10.1002/jso.21095. PMID:18537151
- Lautenschlaeger T, George A, Klimowicz AC, Efstathiou JA, Wu CL, Sandler H, Shipley WU, Tester WJ, Hagan MP, Magliocco AM, et al. Bladder preservation therapy for muscle-invading bladder cancers on Radiation Therapy Oncology Group trials 8802, 8903, 9506, and 9706: vascular endothelial growth factor B overexpression predicts for increased distant metastasis and shorter survival. Oncologist. 2013;18:685-6.
- Zhu M, Zhang Q, Wang X, Kang L, Yang Y, Liu Y, et al. Metformin potentiates anti-tumor effect of resveratrol on pancreatic cancer by down-regulation of VEGF-B signaling pathway. Oncotarget 2016.7(51):84190-200. doi:10.18632/oncotarget.12391. PMID:27705937
- Scrofani SD, Fabri LJ, Xu P, Maccarone P, Nash AD. Purification and refolding of vascular endothelial growth factor-B. Protein Sci. 2000;9:2018-25. doi:10.1110/ps.9.10.2018. PMID:11106176
- Yu L, Liang XH, Ferrara N. Comparing protein VEGF inhibitors: In vitro biological studies. Biochem Biophys Res Commun. 2011;408:276-81. doi:10.1016/j.bbrc.2011.04.014. PMID:21501594
- Gerber HP, Wu X, Yu L, Wiesmann C, Liang XH, Lee CV, Fuh G, Olsson C, Damico L, Xie D, et al. Mice expressing a humanized form of VEGF-A may provide insights into the safety and efficacy of anti-VEGF antibodies. Proc Natl Acad Sci U S A. 2007;104:3478-83. doi:10.1073/pnas.0611492104. PMID:17360669
- Larsen AK, Ouaret D, El Ouadrani K, Petitprez A. Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol Ther. 2011;131:80-90. doi:10.1016/j.pharmthera.2011.03.012. PMID:21439312