
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects nerve cells in the brain and the spinal cord. In a recent study by Steinberg and colleagues, 2 recessive missense mutations were identified in the Cav3.2 T-type calcium channel gene (CACNA1H), in a family with an affected proband (early onset, long duration ALS) and 2 unaffected parents. We have introduced and functionally characterized these mutations using transiently expressed human Cav3.2 channels in tsA-201 cells. Both of these mutations produced mild but significant changes on T-type channel activity that are consistent with a loss of channel function. Computer modeling in thalamic reticular neurons suggested that these mutations result in decreased neuronal excitability of thalamic structures. Taken together, these findings implicate CACNA1H as a susceptibility gene in amyotrophic lateral sclerosis.
Introduction
Amyotrophy lateral sclerosis (ALS) is a progressive neurodegenerative disease affecting motor nerve cells in the brain and in the spinal cord.Citation1 About 10% of ALS is classified as inherited, whereas the remaining 90% of cases appear to occur randomly throughout the population and are considered to be sporadic in nature.Citation2 Thanks to recent advances in sequencing and genotyping technologies, a number of genes underlying familial ALS have been identified and now are associated with about 2-thirds of inherited cases.Citation3 In contrast, the cause of sporadic ALS is not well understood, but may be due to a combination of environmental and genetic risk factors.Citation4 De novo mutations of FUS,Citation5,6 SOD1,Citation7 ERBB4Citation8 and ATXN2Citation9 have been described in sporadic ALS cases. In addition, one form of inheritance that can be misinterpreted as an apparent sporadic form of ALS can arise from homozygous or compound heterozygous recessive variants. In a recent whole exome sequence analysis of case-unaffected-parents trios,Citation10 2 compound heterozygous recessive missense mutations in CACNA1H were identified in a 39-year-old man diagnosed with a early onset (aged 27 years), long-duration variant of ALS. He presented with a slowly progressive upper motor neuron hypertonia and lower motor neuron weakness involving all limbs, as well as dysarthria. Lower motor neuron signs were more prominent in the upper limbs. Widespread acute and chronic denervation was shown on electromyography. A genetic test for Kennedy's disease was negative. There was no history of any neuromuscular disorder, early deaths, or epilepsy in his siblings, parents, grandparents, or aunts and uncles. The c.1215267G>A and c.1215315G>A mutations in CACNA1H caused the substitution of a valine to a methionine (p.V1689M) and of an alanine to a threonine (p.A1705T) in the voltage-gated Cav3.2 T-type calcium channel.
T-type channels are low-voltage-gated calcium channels and are perfectly suited to operate near the resting electrical membrane potential of nerve cells. They support a “window current” providing an incentive for Ca2+ entry,Citation11 and contributing to the resting membrane potential of neurons.Citation12 Additionally, T-type channels play an essential role in regulating neuronal excitability. Their hyperpolarization-induced recovery from inactivation supports rebound burst of action potentials that contribute to various form of neuronal rhythmogenesis.Citation13-16 Their implication in neuronal excitability is further supported by their ability to associate with, and regulate the activity of, calcium-activated and voltage-gated potassium channels, which in turn shape firing properties of nerve cells.Citation17-20 Finally, T-type channels also support a low-threshold form of exocytosis by virtue of their biochemical coupling with vesicular release machinery.Citation21,22 All of these aspects are particularly important to the functioning of the thalamocortical network, a brain structure implicated in motor-related activities.Citation23-26
In the present study, we analyzed the functional expression of the 2 ALS-associated missense mutations in Cav3.2 channels. We show that both mutations produce significant alterations on the channel activity, consistent with a loss of channel function. Computational modeling of the firing properties of thalamic reticular neurons expressing Cav3.2 channel variants revealed a decreased neuronal excitability. Taken together, our findings implicate for the first time CACNA1H as a susceptibility gene in one form of ALS.
Results
Expression of Cav3.2 channel variants
The transmembrane topology of the Cav3.2 pore-forming subunit of T-type channels consists of 4 homologous domains (I to IV), each of them containing 6 putative transmembrane helices (S1 to S6), plus a re-entrant loop (P-loop) that forms the pore of the channel. The two ALS-associated missense mutations, V1689M and A1705T, identified in a case-unaffected-parents trio (), are both located in the fourth membrane domain of the channel, in the segment S3 and in the short extracellular linker connecting segments S3 and S4, respectively (). These mutations affect residues that are highly conserved among T-type channel paralogs and orthologs (), suggesting an important role for these residues in channel function. The two ALS mutations were introduced independently in the human Cav3.2 channel (hCav3.2) and expressed in tsA-201 cells, and expression of the channel variants was confirmed by western blot analysis (). To assess the functional expression of the Cav3.2 mutations, we performed patch-clamp recordings on tsA-201 cells expressing the channel variants. Representative current traces in response to 150 ms depolarizing steps to values ranging between −90 mV and +30 mV, from a holding potential of −100 mV, are shown in for wild-type (WT) hCav3.2 channels, and V1689M and A1705T variants. T-type currents recorded from cells expressing the V1689M variant were found to be significantly reduced compared to cells expressing the WT channel ( and ). For instance, in response to a depolarizing pulse to −30 mV, the mean peak current density was decreased by 30% (p = 0.0002) in cells expressing the V1689M variant (−35.5 ± 2.2 pA/pF, n = 36) compared to cells expressing the WT channel (−51.0 ± 3.2 pA/pF, n = 36). The maximal conductance was reduced by 27% (p = 0.0004) in hCav3.2V1689M-expressing cells (731 ± 39 pS/pF, n = 36) compared to cells expressing the WT channel (996 ± 59 pS/pF, n = 36) (). In contrast, the maximal T-type conductance measured in cells expressing the A1705T channel variant was unaltered (976 ± 62 pS/pF, n = 38) compared to cells expressing the WT channel. To determine whether the impaired T-type current in cells expressing the V1689M variant was caused by an alteration of the channel activity or due to reduced expression at the plasma membrane, we performed surface immunostaining on tsA-201 cells expressing exofacial hemagglutinin (HA)-tagged hCav3.2 (HA-hCav3.2) variants. The expression of HA-hCav3.2 was quantified from low-magnification confocal images obtained from non-permeabilized and permeabilized cells to assess surface and total expression of the channel, respectively (). The surface and total expression levels were similar between the Cav3.2 channel variants, supporting the notion that the V1689M mutation may alter the functioning of the Cav3.2 channel.
Figure 1. Segregation of V1689M and A1705T hCav3.2 mutations in an ALS case-unaffected-parents trio. (A) Pedigree chart of the ALS case-unaffected-parents trio. Filled and open symbols represent affected and unaffected individuals, respectively. (B) Schematic representation of the Cav3.2 channel showing its organization in 4 membrane domains, each containing 6 transmembrane segments. The V1689M mutation (red) is located in the third transmembrane segment of the domain IV. The A1705T mutation (blue) is located in the extracellular linker connecting segments S3 and S4 of domain IV. (C) Alignment of orthologs and (D) paralogs showing that the valine 1689 (V1689) and the arginine 1705 (A1705) are highly conserved across all species and T-type channel isoforms.
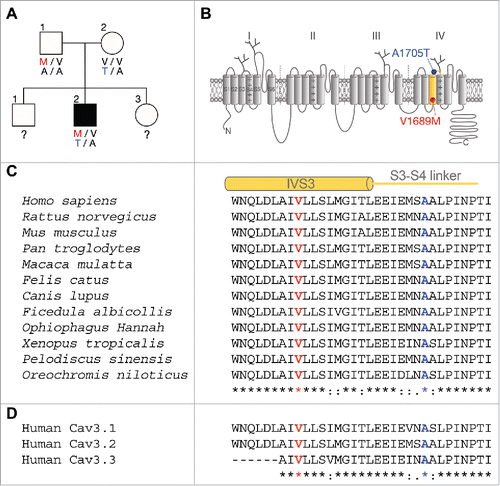
Figure 2. Expression of ALS-associated hCav3.2 variants in tsA-201 cells. (A) Immunoblot of HA-hCav3.2 variants expressed in tsA-201 cells. (B) Representative Ba2+ current traces recorded in response to 150 ms depolarization steps to values ranging between −90 mV and +30 mV, from a holding potential of −100 mV, for wild-type hCav3.2 and V1689M and A1705T channel variants. (C) Corresponding mean current-voltage relationship for hCav3.2 WT (black circles), V1689M (red circles), and A1705T channels (blue circles). (D) Corresponding mean maximal macroscopic Ba2+ conductance. (E) Low magnification confocal images of non-permeabilized (upper panels) and permeabilized (lower panels) tsA-201 cells expressing HA-tagged hCav3.2 channel variants, and stained for HA-hCav3.2 (green) using a primary anti-HA antibody.
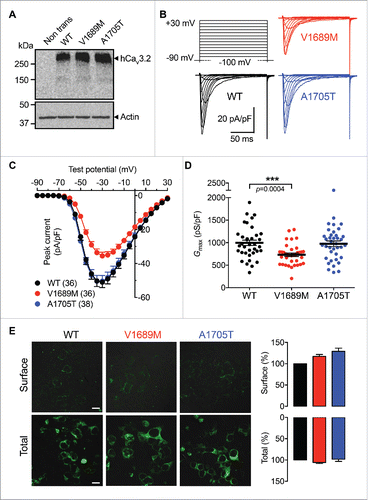
Table 1. Electrophysiological properties of human Cav3.2 channel variants expressed in tsA-201 cells. Activation and inactivation kinetic values are shown at −40 mV.
The V1689M mutation alters activation of hCav3.2 channels
To further assess the impact of ALS-associated CACNA1H mutations on hCav3.2 channel activity, we analyzed the activation properties of hCav3.2 channel variants. The voltage-dependence of the T-type current activation was determined in cells expressing the WT hCav3.2 channel and in cells expressing the ALS channel variants ( and ). The mean half activation potential was shifted toward more positive potentials by +3.0 mV (p<0.0001) in cells expressing the V1689M variant (−42.0 ± 0.5 mV; n = 36) compared to cells expressing the WT channel (−45.0 ± 0.4 mV; n=36). In contrast, the mean half activation potential of the T-type current measured in cells expressing the A1705T variant remained unaltered (−46.1 ± 0.4 mV, n = 38). The activation kinetics of the hCav3.2 channel variants were analyzed by fitting the developing phase of the T-type current with a single exponential function. We found that the T-type current activation was accelerated in cells expressing the hCav3.2 channel mutants ( and ). For instance, in response to a depolarizing step to −40 mV, the time constant of activation of the T-type current recorded from cells expressing the channel mutants was on average 1.2-fold faster (p = 0.0112) in V1689M-expressing cells (3.9 ± 0.1 ms; n = 35) and 1.3-fold faster (p = 0.0002) in A1705T-expressing cells (3.6 ± 0.1 ms; n = 34) than in cells expressing the WT channel (4.6 ± 0.2 ms; n = 36). However, this effect remained marginal, and appeared to be only significant at the lowest voltages tested.
Figure 3. Activation properties of ALS-associated hCav3.2 variants. (A) Mean normalized voltage-dependence of the macroscopic Ba2+ conductance recorded from wild-type hCav3.2 (black circles), V1689M (red circles), and A1705T mutant (blue circles) channel-expressing cells. (B) Corresponding mean half activation potential. (C) Time constant of current activation for WT and ALS-associated hCav3.2 mutants. Left panel shows the first 50 ms of representative current traces for WT (red), V1689M (red), and A1705T (blue) channel variants recorded in response to 150 ms depolarization step to −40 mV from a holding potential of −100 mV.
Altogether, these data indicate a loss of function of hCav3.2 channels carrying the V1689M evidenced by decreased voltage sensitivity, indicating that stronger depolarizations are required to open the channel.
The A1705T mutation alters inactivation of hCav3.2 channels
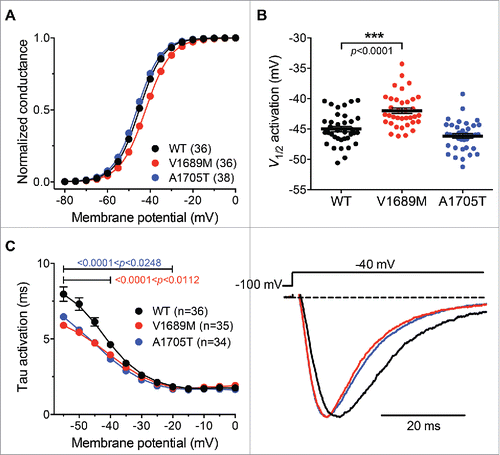
We further investigated the impact of the 2 ALS mutations on the inactivation properties of hCav3.2 channels. The steady state inactivation of hCav3.2 variants was analyzed. The mean normalized voltage-dependence of steady-state inactivation is shown in for WT and ALS hCav3.2 channel variants. The mean half-steady state inactivation potential was shifted toward more negative potentials by −2.1 mV (p = 0.0003) in cells expressing the A1705T variant (−77.1 ± 0.4 mV; n = 23) compared to cells expressing the WT channel (−75.0 ± 0.5 mV; n=23) ( and ). In contrast, the mean half-steady-state inactivation potential of the V1689M channel variant remained unaltered (−74.9 ± 0.6 mV; n = 19). The inactivation kinetics of the hCav3.2 channel variants were analyzed by fitting the decay phase of the T-type current with a single exponential function. There was no significant difference in the inactivation time constant between the WT hCav3.2 and the ALS channel variants ( and ). Finally, the ALS-associated hCav3.2 mutations had no significant effect on the recovery from short-term inactivation ( and ).
Figure 4. Inactivation properties of ALS-associated hCav3.2 variants. (A) Mean normalized voltage-dependence of steady state inactivation for wild-type hCav3.2 (black circles), V1689M (red circles), and A1705T channel variants (blue circles). (B) Corresponding mean half-steady-state inactivation potentials. (C) Time constant of current activation for WT and ALS-associated hCav3.2 mutants. (D) Recovery from short-term inactivation according to a 2-paired-pulse protocol, shown in inset.
Collectively, these results indicate that A1705T specifically produces a negative shift of the voltage-dependence of steady state inactivation of hCav3.2 channels, which ultimately results in decreased channel availability at typical neuronal resting membrane potentials.
The ALS-associated CACNA1H mutations decrease firing of thalamic reticular neurons
T-type channels are highly expressed in the thalamus, especially in reticular thalamic neurons (nRT),Citation27 where they play an essential role in regulating neuronal excitability. To evaluate the impact of ALS-associated CACNA1H mutations on neuronal excitability, we used electrophysiological parameters of Cav3.2 channel variants obtained experimentally, and computational modeling, to simulate the firing of nRT neurons. Since the Cav3.2 channel variants showed significant differences in the steady-state activation, steady state inactivation, and conductance properties, these parameters were introduced into the model. In addition, and because the 2 mutations are found on different alleles, the total T-type current was modeled by the average of the V1689M and A1705T currents to mimic the co-expression of the 2 channel variants in the ALS proband. Consistent with a loss of channel function, computational simulation revealed a decreased firing of nRT neurons co-expressing the V1689M and A1705T channel variants compared to neurons expressing the wild-type channel (). The decreased cellular excitability is evidenced by progressive spike latency and decreased number of spikes in heterozygous V1689M/A1705T neurons (, brown trace) compared to homozygous WT neurons (, black trace and superimposed gray traces). In contrast, and consistent with the absence of neurological symptoms in heterozygous unaffected parents carrying only one of the channel variants, the firing properties of heterozygous V1689M/WT and A1705T/WT nRT neurons were mildly altered. For instance, only a firing latency without spike reduction was observed in V1689M/WT neurons (, red trace), whereas the firing of A1705T/WT neurons (, blue trace) was virtually identical to the firing of homozygous WT neurons.
Figure 5. ALS-associated hCav3.2 mutations alter firing of thalamic neurons. Computational simulations of the firing of nRT neurons for homozygous WT (black), heterozygous V1689M/WT (red) and A1705T/WT (blue), and heterozygous V1689M/A1705T conditions (brown). For each condition the homozygous WT is superimposed in gray for comparison. The current-clamp responses were elicited by 0.0418 nA current injection for 200 ms in a dendrite (dend1[0](0)) of the 3-compartment cell model, from a holding potential of −70 mV.
) of the 3-compartment cell model, from a holding potential of −70 mV.](/cms/asset/46630a97-2fe9-421c-b7c5-3d581cd502f4/kchl_a_1204497_f0005_c.gif)
Given the importance of thalamocortical network activity in the control of motor-related functions, alteration of reticular thalamic neuron firing caused by ALS-associated Cav3.2 channel variants represents an important mechanism that could account for the development of the disease. In addition, it provides a functional explanation for the absence of neurological symptoms in case-unaffected-heterozygous-parents carrying only one mutation compared to the proband carrying the 2 channel variants.
Discussion
Although 90% of ALS cases are considered to be sporadic because of their random appearance in the population, one form of inheritance that can be misinterpreted as a non-genetic form of ALS can arise from recessive variants. In the present study, we have analyzed the functional impact of 2 recessive mutations in CACNA1H, identified in a case-unaffected-parents trio, on the functioning of Cav3.2 channels. The two mutations, when expressed in tsA-201 cells, caused mild but significant changes to Cav3.2 activity. Consistent with a loss of channel function, computational modeling revealed decreased firing of thalamic neurons that expressed ALS-associated Cav3.2 channel variants.
The V1689M and A1705T Cav3.2 mutations were located in the domain IV of the channel, in the third transmembrane segment (S3) and in the short extracellular linker connecting the third and fourth transmembrane segments (S3-S4), respectively. Consistent with the notion that these mutations affect residues that are important for the functioning of the channel, patch clamp recordings in cells expressing the ALS-associated Cav3.2 variants revealed a depolarizing shift of the voltage dependence of steady-state of activation caused by the V1689M mutation, and a hyperpolarizing shift of the voltage dependence of the steady state inactivation induced by the A1705T mutation. Interestingly, the A1705T mutation was previously reported in a patient with myoclonic-astatic epilepsy and febrile seizures, and initial biophysical analysis revealed a mild but significantly faster recovery from short-term inactivation when the mutation was introduced into the short Cav3.2 channel isoform (i.e., which does not contain the exon 25 located encoding for a short region of the cytoplasmic III-IV linker).Citation28 In our study we used the long Cav3.2 channel variant (i.e., containing the exon 25), which may account for the biophysical difference between the 2 studies. It is also important to note that the A1705T variant is relatively frequent in non-affected individuals. In contrast, the V1689M appears very rarely, suggesting that this particular variant may be highly associated with ALS.
Interestingly, while the 2 ALS-associated mutations produced distinct effects on the channel properties, they both translated into a loss of channel function. Consistent with the expression of Cav3.2 channels in thalamic brain structures, and their role in generating low-threshold calcium potentials and burst firing, computational modeling revealed decreased firing of nRT neurons expressing a combination of ALS-associated Cav3.2 channel variants. This notion is consistent with previous studies using advance brain imaging techniques that showed decreased activity in various brain regions, including the thalamus in ALS patients.Citation29-34 Considering that a number of thalamic nuclei project their axons to motor cortical areas,Citation35 alteration of thalamic network activity caused by ALS-associated mutations may alter the electrical input received by motor neuronal structures, which may eventually cause an alteration of motor movement control. Alteration in T-type channel resulting in a loss of function can also alter the balance between excitatory and inhibitory neuronal networks. Finally, given the importance of T-type channels in modulating other ion channel conductances,Citation17 it is conceivable that the phenotypic expression of a particular variant may depend on its interaction with other ion channels, such as calcium-activated and voltage-gated potassium channels.
The notion that ALS-associated CACNA1H variants produce relatively mild changes on channel activity is in line with previous studies reporting discrete biophysical alterations of Cav3.2 channels that carry mutations associated with idiopathic generalized epilepsiesCitation28,36-39 and chronic pain.Citation40 While these variants may not be sufficiently pathogenic to cause the disease on their own, in combination with other variants or environmental factors they could increase disease susceptibility. This may also explain the absence of neurological phenotype in the parents of the individual, who each carry only one of the CACNA1H variants which according to our simulation experiment is not sufficient to significantly alter neuronal excitability. Of note, and consistent with the relatively mild alterations cause by CACNA1H mutations, the patient reported here showed the first signs of weakness when he was at the relatively young age of 27 y old, and the disease progressed only slowly, both features unusual in classic ALS.
In conclusion, we reported the first functional characterization of Cav3.2 channel mutations associated with ALS, establishing for the first time CACNA1H as a susceptibility gene in ALS. Our electrophysiological study and computational modeling are consistent with decreased channel activity and neuronal excitability in brain structures important for controlling motor-related functions. These mutations and their functional changes may act synergistically with other factors, all of which could contribute to susceptibility to ALS.
Material and methods
Plasmids cDNA constructs and site-directed mutagenesis
The human wild-type (WT) HA-tagged Cav3.2 (HA-hCav3.2WT) in pcDNA3.1 was used as a template for insertion of V1689M and A1705T missense mutations. Site-directed mutagenesis using overlapping extension PCR method was performed using the following external primers: 5′-GGATCCCAAGCTTGGTACCG-3′ (forward) and 5′-CTTCATGGCCCTCTAGAGGATCC-3′ (reverse); and the following mutagenic internal primers: V1689M: 5′- GGCCATCATGCTGCTGTCACTCATGGGCATC -3′ (forward) and 5′- CAGCAGCATGATGGCCAGGTCCAGCTGGTTC -3′ (reverse); A1705T: 5′- GATGAGCACCGCGCTGCCCATCAACC -3′ (forward) and 5′- CAGCGCGGTGCTCATCTCTATCTCCTCCAGCG -3′ (reverse). Mutated PCR products were inserted into HindIII/XbaI sites of pcDNA3.1-HA-hCav3.2 vector to generate the respective mutated hCav3.2V1689M and hCav3.2A1705M channel constructs. All final constructs were verified by sequencing of the full-lengh cDNAs.
Cell culture and heterologous expression
Human embryonic kidney tsA-201 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen), and maintained under standard conditions at 37°C in a humidified atmosphere containing 5 % CO2. Cells were transfected with plasmid cDNAs encoding Cav3.2 channel variants using the calcium phosphate method.
Patch-clamp electrophysiology
Patch-clamp recordings were performed 72 h after transfection in the whole-cell configuration of the patch-clamp technique at room temperature (22–24°C) in a bath solution containing (in millimolar): 5 BaCl2, 5KCl, 1 MgCl2, 128 NaCl, 10 TEA-Cl, 10 D-glucose, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH7.2 with NaOH). Patch pipettes had a resistance of 2–4 MΩ when filled with a solution containing (in millimolar): 110 CsCl, 3 Mg-ATP, 0.5 Na-GTP, 2.5 MgCl2, 5 D-glucose, 10 EGTA, and 10 HEPES (pH7.4 with CsOH). Whole-cell patch-clamp recordings were performed using an Axopatch 200B amplifier (Axon Instruments). Acquisition and analysis were performed using pClamp 10 and Clampfit 10 software, respectively (Axon Instruments).
Ba2+ currents were recorded in response to 150 ms-long depolarizing to various potentials applied every 5 s from a holding potential of −100 mV. The linear leak component of the current was corrected online and current traces were digitized at 10 kHz, and filtered at 2 kHz. The voltage dependence of the peak Ba2+ current density was fitted with the following modified Boltzmann equation:with I(V) being the peak current amplitude at the command potential V, Gmax the maximum conductance, Vrev the reversal potential, V0.5 the half-activation potential, and k the slope factor. The voltage dependence of the whole-cell Ba2+ conductance was calculated using the following modified Boltzmann equation:
with G(V) being the Ba2+ conductance at the command potential V.
The steady-state voltage-dependence of inactivation of the Ba2+ current was determined by measuring the peak current amplitude in response to a 150 ms-long depolarizing step to −20 mV applied after a 5 s-long conditioning prepulse ranging from −105 mV to −50 mV. The current amplitude obtained during each test pulse was normalized to the maximum at −105 mV and plotted as a function of the prepulse potential. The voltage-dependence of the steady state inactivation was fitted with the following 2-state Boltzmann function:with Imax corresponding to the maximal peak current amplitude and V0.5 to the half-inactivation voltage.
The recovery from inactivation was studied using a double-pulse protocol from a holding potential of −100 mV to ensure complete inactivation of Cav3.2 channels. The cell membrane was depolarized for 2 s to −20 mV (prepulse) to ensure complete channel inactivation and then to −20 mV from 150 ms (test pulse) after an increasing time period (interpulse interval) between 0.5 ms and 8 s. The peak current from the test pulse was plotted as a ratio of the maximum prepulse current versus interpulse interval. The data were fitted with a single-exponential function:where A is the amplitude of the exponential component and τ the time constant.
SDS-PAGE and immunoblot analysis
Total lysates of tsA-201 cells expression HA-tagged Cav3.2 variants were separated on 5-20% gradient SDS-PAGE and transferred onto PVDF membrane (Millipore). For detection of the HA-hCav3.2 channel, the membrane was incubated with a primary rat monoclonal anti-HA antibody (Roche) diluted at 1:1000 for one hour at room temperature, washed in PBS / Tween-20 buffer, and incubated with a secondary HRP-conjugated antibody (Jackson ImmunoResearch) diluted at 1:10,000. Immunoreactive bands were detected by enhanced chemiluminescence.
Surface immunostaining
Twenty-four hours before the experiment, cells expressing HA-hCav3.2 channels were seeded on poly-L-lysine-coated glass coverslips. Cells were incubated for 30 min at 37°C with a primary monoclonal mouse anti-HA antibody (Abcam) diluted in DMEM at 1:1000, washed with PBS, fixed for 7 min in 4 % paraformaldehyde, and blocked for 45 min in blocking buffer (5 % FBS in PBS). Cells were then incubated for 1 h at room temperature with a secondary goat polyclonal anti-mouse Alexa488-conjugated antibody (Jackson ImmunoResearch) diluted in blocking buffer at 1:1000, washed, and mounted on microscope glass slides with ProLong Gold mounting medium (Life Technologies). Confocal images were acquired at low magnification with a Zeiss LSM780 microscope and the field fluorescence intensity was analyzed using ImageJ software. In order to visualize intracellular channels, cells were permeabilized with 0.2 % Triton X-100 for 10 min before incubation with the secondary antibody.
Computational modeling
Simulation of thalamic reticular neuron (nRT) firing was performed using the NEURON simulation environment (https://senselab.med.yale.edu/modeldb).Citation41 The electrophysiological properties of the wild-type (WT), V1689M, and A1705T Cav3.2 channel variants were modeled using Hodgkin-Huxley equations as previously describedCitation42 and introduced into a nRT neuron model.Citation43 Only the steady-state activation and inactivation properties were changed. For the simulation of the V1689M variant, the maximal conductance was multiplied by 0.73 to take into account the additional decreased conductance of this variant. Simulation of heterozygous conditions was performed by defining the total T-type current as the average of the 2 channel variants or the average of one channel variant and one WT channel.
Statistical analysis
Data values are presented as mean ± SEM for n experiments. Statistical significance was determined using Student's unpaired t test: *p < 0.05, **p < 0.01, ***p < 0.001, and NS: statistically not different.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank all family members for their contribution to this study. We are grateful to Dr. Gerald W. Zamponi (University of Calgary, Canada) for his valuable comments on our manuscript.
Funding
Work in the Weiss laboratory is supported by the Czech Science Foundation (grant 15-13556S), the Czech Ministry of Education Youth and Sports (grant 7AMB15FR015), and the Institute of Organic Chemistry and Biochemistry (IOCB). Y.R. and J.L. are supported by an IOCB postdoctoral fellowship.
References
- Salameh JS, Brown RH, Berry JD. Amyotrophic lateral sclerosis: review. Semin Neurol 2015; 35:469-76; PMID:26502769; http://dx.doi.org/10.1055/s-0035-1558984
- Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet 2011; 377:942-55; PMID:21296405; http://dx.doi.org/10.1016/S0140-6736(10)61156-7
- Renton AE, Chi∫ A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014; 17:17-23; PMID:24369373; http://dx.doi.org/10.1038/nn.3584
- Morahan JM, Yu B, Trent RJ, Pamphlett R. Genetic susceptibility to environmental toxicants in ALS. Am J Med Genet B Neuropsychiatr Genet 2007; 144B:885-90; PMID:17503480; http://dx.doi.org/10.1002/ajmg.b.30543
- Chi∫ A, Calvo A, Moglia C, Ossola I, Brunetti M, Sbaiz L, Lai SL, Abramzon Y, Traynor BJ, Restagno G. A de novo missense mutation of the FUS gene in a “true” sporadic ALS case. Neurobiol Aging 2011; 32:553.e23-6; PMID:Can't
- DeJesus-Hernandez M, Kocerha J, Finch N, Crook R, Baker M, Desaro P, Johnston A, Rutherford N, Wojtas A, Kennelly K, et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat 2010; 31:E1377-89; PMID:20232451; http://dx.doi.org/10.1002/humu.21241
- Alexander MD, Traynor BJ, Miller N, Corr B, Frost E, McQuaid S, Brett FM, Green A, Hardiman O. True” sporadic ALS associated with a novel SOD-1 mutation. Ann Neurol 2002; 52:680-3; PMID:12402272; http://dx.doi.org/10.1002/ana.10369
- Takahashi Y, Fukuda Y, Yoshimura J, Toyoda A, Kurppa K, Moritoyo H, Belzil VV, Dion PA, Higasa K, Doi K, et al. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am J Hum Genet 2013; 93:900-5; PMID:24119685; http://dx.doi.org/10.1016/j.ajhg.2013.09.008
- Laffita-Mesa JM, Rodríguez Pupo JM, Moreno Sera R, Vázquez Mojena Y, Kourí V, Laguna-Salvia L, Martínez-Godales M, Valdevila Figueira JA, Bauer PO, Rodríguez-Labrada R, et al. De novo mutations in ataxin-2 gene and ALS risk. PLoS One 2013; 8:e70560; PMID:23936447; http://dx.doi.org/10.1371/journal.pone.0070560
- Steinberg KM, Yu B, Koboldt DC, Mardis ER, Pamphlett R. Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep 2015; 5:9124; PMID:25773295; http://dx.doi.org/10.1038/srep09124
- Crunelli V, Tóth TI, Cope DW, Blethyn K, Hughes SW. The ‘window’ T-type calcium current in brain dynamics of different behavioural states. J Physiol 2005; 562:121-9; PMID:15498803; http://dx.doi.org/10.1113/jphysiol.2004.076273
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. J Neurosci 2010; 30:99-109; PMID:20053892; http://dx.doi.org/10.1523/JNEUROSCI.4305-09.2010
- Crunelli V, Cope DW, Hughes SW. Thalamic T-type Ca2+ channels and NREM sleep. Cell Calcium 2006; 40:175-90; PMID:16777223; http://dx.doi.org/10.1016/j.ceca.2006.04.022
- Bal T, McCormick DA. Synchronized oscillations in the inferior olive are controlled by the hyperpolarization-activated cation current I(h). J Neurophysiol 1997; 77:3145-56; PMID:9212264
- Beurrier C, Congar P, Bioulac B, Hammond C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J Neurosci 1999; 19:599-609; PMID:9880580
- Sotty F, Danik M, Manseau F, Laplante F, Quirion R, Williams S. Distinct electrophysiological properties of glutamatergic, cholinergic and GABAergic rat septohippocampal neurons: novel implications for hippocampal rhythmicity. J Physiol 2003; 551:927-43; PMID:12865506; http://dx.doi.org/10.1113/jphysiol.2003.046847
- Turner RW, Zamponi GW. T-type channels buddy up. Pflugers Arch 2014; 466:661-75; PMID:24413887; http://dx.doi.org/10.1007/s00424-013-1434-6
- Anderson D, Mehaffey WH, Iftinca M, Rehak R, Engbers JD, Hameed S, Zamponi GW, Turner RW. Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat Neurosci 2010; 13:333-7; PMID:20154682; http://dx.doi.org/10.1038/nn.2493
- Rehak R, Bartoletti TM, Engbers JD, Berecki G, Turner RW, Zamponi GW. Low voltage activation of KCa1.1 current by Cav3-KCa1.1 complexes. PLoS One 2013; 8:e61844; PMID:23626738; http://dx.doi.org/10.1371/journal.pone.0061844
- Anderson D, Engbers JD, Heath NC, Bartoletti TM, Mehaffey WH, Zamponi GW, Turner RW. The Cav3-Kv4 complex acts as a calcium sensor to maintain inhibitory charge transfer during extracellular calcium fluctuations. J Neurosci 2013; 33:7811-24; PMID:23637173; http://dx.doi.org/10.1523/JNEUROSCI.5384-12.2013
- Weiss N, Hameed S, Fernández-Fernández JM, Fablet K, Karmazinova M, Poillot C, Proft J, Chen L, Bidaud I, Monteil A, et al. A Ca(v)3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J Biol Chem 2012; 287:2810-8; PMID:22130660; http://dx.doi.org/10.1074/jbc.M111.290882
- Weiss N, Zamponi GW, De Waard M. How do T-type calcium channels control low-threshold exocytosis. Commun Integr Biol 2012; 5:377-80; PMID:23060963; http://dx.doi.org/10.4161/cib.19997
- Klockgether T, Schwarz M, Turski L, Sontag KH. The rat ventromedial thalamic nucleus and motor control: role of N-methyl-D-aspartate-mediated excitation, GABAergic inhibition, and muscarinic transmission. J Neurosci 1986; 6:1702-11; PMID:2872282
- Sommer MA. The role of the thalamus in motor control. Curr Opin Neurobiol 2003; 13:663-70; PMID:14662366; http://dx.doi.org/10.1016/j.conb.2003.10.014
- Prevosto V, Sommer MA. Cognitive control of movement via the cerebellar-recipient thalamus. Front Syst Neurosci 2013; 7:56; PMID:24101896; http://dx.doi.org/10.3389/fnsys.2013.00056
- Bosch-Bouju C, Hyland BI, Parr-Brownlie LC. Motor thalamus integration of cortical, cerebellar and basal ganglia information: implications for normal and parkinsonian conditions. Front Comput Neurosci 2013; 7:163; PMID:24273509; http://dx.doi.org/10.3389/fncom.2013.00163
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci 1999; 19:1895-911; PMID:10066243
- Heron SE, Khosravani H, Varela D, Bladen C, Williams TC, Newman MR, Scheffer IE, Berkovic SF, Mulley JC, Zamponi GW. Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann Neurol 2007; 62:560-8; PMID:17696120; http://dx.doi.org/10.1002/ana.21169
- Foerster BR, Welsh RC, Feldman EL. Twenty-five years of neuroimaging in amyotrophic lateral sclerosis. Nat Rev Neurol 2013; 9:513-24; PMID:23917850; http://dx.doi.org/10.1038/nrneurol.2013.153
- Sach M, Winkler G, Glauche V, Liepert J, Heimbach B, Koch MA, Büchel C, Weiller C. Diffusion tensor MRI of early upper motor neuron involvement in amyotrophic lateral sclerosis. Brain 2004; 127:340-50; PMID:14607785; http://dx.doi.org/10.1093/brain/awh041
- Sharma KR, Saigal G, Maudsley AA, Govind V. 1H MRS of basal ganglia and thalamus in amyotrophic lateral sclerosis. NMR Biomed 2011; 24:1270-6; PMID:21404355; http://dx.doi.org/10.1002/nbm.1687
- Sudharshan N, Hanstock C, Hui B, Pyra T, Johnston W, Kalra S. Degeneration of the mid-cingulate cortex in amyotrophic lateral sclerosis detected in vivo with MR spectroscopy. AJNR Am J Neuroradiol 2011; 32:403-7; PMID:21087934; http://dx.doi.org/10.3174/ajnr.A2289
- Sharma KR, Sheriff S, Maudsley A, Govind V. Diffusion tensor imaging of basal ganglia and thalamus in amyotrophic lateral sclerosis. J Neuroimaging 2013; 23:368-74; PMID:22273090; http://dx.doi.org/10.1111/j.1552-6569.2011.00679.x
- Stoppel CM, Vielhaber S, Eckart C, Machts J, Kaufmann J, Heinze HJ, Kollewe K, Petri S, Dengler R, Hopf JM, et al. Structural and functional hallmarks of amyotrophic lateral sclerosis progression in motor- and memory-related brain regions. Neuroimage Clin 2014; 5:277-90; PMID:25161894; http://dx.doi.org/10.1016/j.nicl.2014.07.007
- Jones EG. Thalamic circuitry and thalamocortical synchrony. Philos Trans R Soc Lond B Biol Sci 2002; 357:1659-73; PMID:12626002; http://dx.doi.org/10.1098/rstb.2002.1168
- Chen Y, Lu J, Pan H, Zhang Y, Wu H, Xu K, Liu X, Jiang Y, Bao X, Yao Z, et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol 2003; 54:239-43; PMID:12891677; http://dx.doi.org/10.1002/ana.10607
- Khosravani H, Altier C, Simms B, Hamming KS, Snutch TP, Mezeyova J, McRory JE, Zamponi GW. Gating effects of mutations in the Cav3.2 T-type calcium channel associated with childhood absence epilepsy. J Biol Chem 2004; 279:9681-4; PMID:14729682; http://dx.doi.org/10.1074/jbc.C400006200
- Khosravani H, Bladen C, Parker DB, Snutch TP, McRory JE, Zamponi GW. Effects of Cav3.2 channel mutations linked to idiopathic generalized epilepsy. Ann Neurol 2005; 57:745-9; PMID:15852375; http://dx.doi.org/10.1002/ana.20458
- Peloquin JB, Khosravani H, Barr W, Bladen C, Evans R, Mezeyova J, Parker D, Snutch TP, McRory JE, Zamponi GW. Functional analysis of Ca3.2 T-type calcium channel mutations linked to childhood absence epilepsy. Epilepsia 2006; 47:655-8; PMID:16529636; http://dx.doi.org/10.1111/j.1528-1167.2006.00482.x
- Souza IA, Gandini MA, Wan MM, Zamponi GW. Two heterozygous Cav3.2 channel mutations in a pediatric chronic pain patient: recording condition-dependent biophysical effects. Pflugers Arch 2016; 468:635-42; PMID:26706850; http://dx.doi.org/10.1007/s00424-015-1776-3
- Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput 1997; 9:1179-209; PMID:9248061; http://dx.doi.org/10.1162/neco.1997.9.6.1179
- Huguenard JR, McCormick DA. Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. J Neurophysiol 1992; 68:1373-83; PMID:1279135
- Destexhe A, Contreras D, Steriade M, Sejnowski TJ, Huguenard JR. In vivo, in vitro, and computational analysis of dendritic calcium currents in thalamic reticular neurons. J Neurosci 1996; 16:169-85; PMID:8613783