
Dysautonomia is a well-established contributor to the development and progression of hypertension and many other cardiovascular diseases. Sympathetic hyperactivity and vagal impairment are features of human hypertension, as well as in subjects with a familial predisposition for hypertension.Citation1,2 This neural phenotype is also observed in the spontaneously hypertensive rat (SHR).Citation3 Moreover, it is widely accepted that autonomic imbalance contributes to the pathogenesis of hypertension itself, where emerging research is beginning to shed light on the key cellular and molecular changes that occur in diseased neurons.Citation3,4
The postganglionic sympathetic stellate neurons (PGSNs) of the SHR that predominantly innervate the heart, display increased intracellular calcium [Ca2+]i transients linked to impaired neuronal nitric oxide synthase (nNOS) activity.Citation3 Together, with downstream reductions in nitric oxide (NO)-cyclic guanosine monophosphate (cGMP), this results in enhanced end-organ neurotransmission.Citation3 Pharmacological and genetic techniques aimed at enhancing NO-cGMP-protein kinase G (PKG) signaling have been successful in rectifying the Ca2+ phenotype in SHR PGSNs and decreasing sympathetic hyperactivity.Citation3 Interestingly, sympathetic impairment is present in young pro-hypertensive SHRs (pro-SHR), suggesting that these intracellular changes form early hallmarks of hypertension; however, the precise nature of events that trigger sympathetic dysfunction remain unclear.
Recently, we published data suggesting that sympathetic hyperactivity in the stellate neurons from the pro-SHR may be triggered by dysregulated Ca2+ channel activity, resulting in greater Ca2+ influx.Citation4 N-type Ca channels (Cav2.2) were identified as the major contributor to Ca2+ entry in PGSNs, that in turn facilitate sympathetic neurotransmission. Inhibition of the N-type Ca2+ channel also reduces the propensity for fatal ventricular arrhythmias and ameliorates autonomic dysfunction in a heart failure mouse model.Citation2 When taken together these findings support a significant physiological role of the N-type Ca2+ channel in neural modulation associated with cardiovascular disease.
In our study we reported that PGSN whole-cell Ca2+ currents of the pro-SHR are greater when compared to normotensive rats.Citation4 Moreover, we demonstrated a novel link between impaired cyclic nucleotide signaling and increased N-type Ca2+ channel activity in pro-hypertension.Citation4 cAMP and cGMP are ubiquitous second messenger cyclic nucleotide signaling molecules that regulate fundamental intracellular processes through direct activation of their respective kinases: protein kinase A (PKA) and PKG. Importantly, cyclic nucleotide regulation of neuronal [Ca2+]i is necessary for normal PGSN function. Indeed, site-specific phospho-regulation of several voltage-gated Ca2+ channel subtypes has been well documented, where a fine balance between PKA and PKG activity is maintained in order to regulate Ca2+-dependent neurotransmission. Additional regulatory mechanisms are also maintained to ensure high signaling fidelity, whereby cAMP and cGMP are able to modulate alternative signaling pathways through the activation and / or inhibition of specific phosphodiesterases (PDEs).Citation5
To test a link between N-type Ca2+ channels and cyclic nucleotide activity we carried out patch-clamp recordings and Förster Resonance Energy Transfer (FRET) microscopy. Pharmacological increases in cGMP were effective in reducing N-Type Ca2+ currents in neurons from the pro-SHR to the same level as that recorded from control PGSNs, demonstrating that cyclic nucleotide regulation of the N-type Ca2+ channels is impaired in prohypertensive states. Real-time FRET recordings of cAMP and PKA illustrated divergent responses to cGMP in PGSNs in health and disease, suggesting significantly altered cyclic nucleotide signaling in diseased neurons. Indeed, in healthy neurons, pharmacological increases in cGMP led to cAMP generation and increased PKA activity. Conversely, in pro-SHR PGSNs, cGMP had no effect on cAMP or PKA signaling. These observations suggest that cross-talk between cyclic nucleotide signaling pathways is impaired in disease, and likely arises as a result of dysregulated PDE activity.Citation4
PDE regulation of cAMP and cGMP cross-talk is considered critically important in maintaining cell-signaling processes and for restricting cyclic nucleotide signaling within specific subcellular microdomains.Citation5 Three families of PDEs (PDE1-3) have affinities for both cAMP and cGMP,Citation5 and recently both PDE2A and PDE3A have been implicated in the pathogenesis of hypertension.Citation6,7 We confirmed the presence of PDE2A and demonstrated pharmacologically a role for PDE3 in PGSNs. Given that PDE2A is activated by cGMP, and PDE3 is inhibited by cGMP, we suggest that the differential cAMP responses to cGMP observed in healthy versus diseased neurons, may arise as a result of altered PDE2/3 activity ().
Figure 1. In healthy neurons, enhancing cGMP increases cAMP and PKA presumably via cGMP-dependent inhibition of PDE3. In pro-hypertensive SHR neurons, cGMP elevation reverses the ICaN phenotype, presumably by enhancing cross-talk between cGMP and cAMP signaling via activation of PDE2A. Red arrows highlight imparied activity in pro-SHR. Yellow arrows depict the downstream effects of pharmacologically enhancing cGMP.
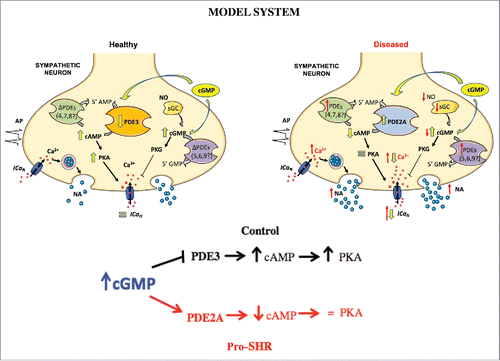
Other interesting but currently unexplored alternative explanations for our observation of impaired cyclic nucleotide cross-talk, might include an increased number of cGMP-specific hydrolysing PDE isoforms in the SHR such as PDE5, 6 or 9; whereby excessive hydrolysis of cGMP in diseased neurons could prevent the activation of cAMP signaling pathways. Notably, PDE9a expression is upregulated in clinical cardiac hypertrophy and heart failure.Citation8 Another possibility is that cAMP-hydrolysing PDE isoforms (e.g. PDE 4, 7, 8) are upregulated in diseased PGSNs, facilitating excessive cAMP hydrolysis following physiological stimuli. However, there is no compelling evidence to date that these PDE isoforms are present in sympathetic neurons. Establishing which PDE isoform's contribute to sympathetic dysfunction and how this impairment arises is one of the key goals for understanding the basis of autonomic-driven metabolic phenotypes. Moreover, pharmacological or genetic targeting of specific PDE isoform's within sympathetic nerves may have significant therapeutic potential for rectifying dysautonomia associated with many cardiovascular diseases.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The Wellcome Trust are the primary funders for this project. The author is funded through the Wellcome Trust as part of the OXION initiative in Ion Channels and Disease at the University of Oxford. The BHF were also secondary funders for the project.
References
- Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Phys Revs 2010; 90(2):513-557; PMID:20393193; https://doi.org/10.1152/physrev.00007.2009
- Yamada Y, Kinoshita H, Kuwahara K, Nakagawa Y, Kuwabara Y, Minami T, Yamada C, Shibata J, Nakao K, Cho K, et al. Inhibition of N-type Ca2+ channels ameliorates an imbalance in cardiac autonomic nerve activity and prevents lethal arrhythmias in mice with heart failure. Cardiovasc Res 2014; 104(1):183-193; PMID:25100767; https://doi.org/10.1093/cvr/cvu185
- Li D, Paterson DJ. Cyclic nucleotide regulation of cardiac sympatho-vagal responsiveness. J Physiol 2016; 594(14):3993-4008; PMID:26915722; https://doi.org/10.1113/JP271827
- Larsen HE, Bardsley EN, Lefkimmiatis K, Paterson DJ. Dysregulation of neuronal Ca2+ channel linked to heightened sympathetic phenotype in prohypertensive states. J. Neurosci 2016; 36(33):8562-73; PMID:27535905; https://doi.org/10.1523/JNEUROSCI.1059-16.2016
- Zaccolo M, Movsesian MA. cAMP and cGMP signaling cross-talk: Role of Phosphodiesterases and implications for cardiac pathophysiology. Circ Res 2007; 100(11):1569-78; PMID:17556670; https://doi.org/10.1161/CIRCRESAHA.106.144501
- Li D, Lu CJ, Hao G, Wright H, Woodward L, Liu K, Vergari E, Surdo NC, Herring N, Zaccolo M, et al. Efficacy of B-type natriuretic peptide is coupled to phosphodiesterase 2A in cardiac sympathetic neurons. Hypertension 2015; 66(1):190-8; PMID:25916722; https://doi.org/10.1161/HYPERTENSIONAHA.114.05054
- Boda H, Uchida H, Takaiso N, Ouchi Y, Fujita N, Kuno A, Hata T, Nagatani A, Funamoto Y, Miyata M, et al. A PDE3A mutation in familial hypertension and brachydactyly syndrome. J Hum Genet 2016; 61(8):701-703; PMID:27053290; https://doi.org/10.1038/jhg.2016.32
- Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo S-H, Danner T, Zhang M, Rainer PP, et al. Phosphodiesterase 9A controls nitric-oxide- independent cGMP and hypertrophic heart disease. Nature 2015; 519(7544):472-476; PMID:25799991; https://doi.org/10.1038/nature14332