
ABSTRACT
The TREK-1 channel, the TWIK-1-related potassium (K+) channel, is a member of a family of 2-pore-domain K+ (K2P) channels, through which background or leak K+ currents occur. An interesting feature of the TREK-1 channel is the run-up of current: i.e. the current through TREK-1 channels spontaneously increases within several minutes of the formation of the whole-cell configuration. To investigate whether intracellular transport is involved in the run-up, we established 293T cell lines stably expressing the TREK-1c channel (K2P2.1) and examined the effects of inhibitors of membrane protein transport, N-methylmaleimide (NEM), brefeldin-A, and an endocytosis inhibitor, pitstop2, on the run-up. The results showing that NEM and brefeldin-A inhibited and pitstop2 facilitated the run-up suggest the involvement of intracellular protein transport. Correspondingly, in cells stably expressing the mCherry-TREK-1 fusion protein, NEM decreased and pitstop2 increased the cell surface localization of the fusion protein. Furthermore, the run-up was inhibited by the intracellular application of a peptide of the C-terminal fragment TREK335–360, corresponding to the interaction site with microtubule-associated protein 2 (Mtap2). This peptide also inhibited the co-immunoprecipitation of Mtap2 with anti-mCherry antibody. The extracellular application of an ezrin inhibitor (NSC668394) also suppressed the run-up and surface localization of the fusion protein. The co-application of these inhibitors abolished the TREK-1c current, suggesting that the additive effects of ezrin and Mtap2 enhance the surface expression of TREK-1c channels and the run-up. These findings clearly showed the involvement of intracellular transport in TREK-1c current run-up and its mechanism.
Potassium(K+)channels play crucial roles in the regulation of the resting membrane potential and excitability of various cells. Among the broad range of K+ channel families, the most recently identified family is one of the 2-pore domain K+ (K2P) channels responsible for background or leak K+ currents.Citation1-4 Each K2P subunit has internal N and C termini, 4 transmembrane domains, and 2 pore-forming loops. The mammalian K2P family now comprises 15 members, one of which is TREK-1 (TWIK-related K+ channel).Citation3 The TREK-1 channel is strongly expressed throughout the nervous system as well as in several non-neuronal tissues and has multiple physiologic and pathological roles.Citation5-8 The TREK-1 channel may be stimulated by several chemical and physical stimuli such as anesthetics, polyunsaturated fatty acids, intracellular acidification, temperature, and mechanical stretch.Citation2,9-13 In contrast, the TREK-1 channel is inhibited by neurotransmitters and hormones that activate G-protein coupled receptors.Citation4,10,14 TREK-1 channel-deficient mice were previously shown to be more vulnerable to epileptic seizures, and ischemia and more resistant to volatile anesthetics.Citation15
To maintain appropriate current levels, K+ channels are strictly regulated in several ways, e.g., the synthesis of mRNAs,Citation16 degradation,Citation17 and intracellular transport to and from the plasma membrane.Citation18 The current of the TREK-1 channel appears to be regulated by intracellular transport, in which the cytosolic C-terminus has been shown to play a key role: the cell surface expression of the TREK channel was enhanced by an interaction between the C-terminal domain and microtubule-associated protein 2 (Mtap2),Citation19 coat protein complex I (β-COP),Citation20 and neurotensin receptor 3 (NTSR3/Sortilin).Citation21 The TREK-1 channel was previously found to co-localize with ezrin and induce the formation of actin-rich protrusions.Citation22
An interesting feature of the TREK-1 channel is the run-up of channel current: i.e., current through TREK-1 channels spontaneously increases within several minutes after whole-cell access.Citation14,23 A previous study demonstrated that the run-up was inhibited by mutations in phosphorylation sites in the C-terminal domain.Citation14 Since C-terminal domain is required for the surface localization of the TREK-1 channel, the run-up might be regulated by transport to the plasma membrane. Here we established 293T cell lines stably expressing TREK-1c channels and showed that intracellular transport of TREK-1c channels to the cell surface and their stabilization in the plasma membrane are major mechanisms responsible for the run-up.
Results
Involvement of intracellular transport of the TREK channel in the run-up
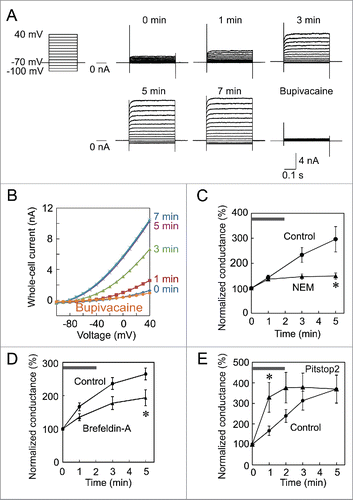
We previously prepared a lentiviral vector that expresses GFP and TREK-1c.Citation24 Using this viral vector, we established the 293T cell line, TR-1, which stably expresses these proteins. We initially investigated whether the TREK-1c current exhibits the run-up in this cell line. Immediately after whole-cell access, small outward currents were recorded in response to step pulses (). These outward currents increased gradually and spontaneously, and reached a maximum level 5 minutes after establishing the whole-cell clamp mode. To confirm that these increasing currents flowed through TREK-1c channels, we applied the selective blocker, bupivacaine,Citation25 and found that the current was almost completely inhibited. The current-voltage relationship showed outward rectification ().
Figure 1. Run-up of the TREK-1c current in a TR-1 cell line and effects of NEM and pitstop2 on the run-up. (A) Run-up of the TREK-1c current. Immediately after whole-cell access, the TREK-1c current was evoked by step-pulses of 400 ms from −100 to 40 mV from a holding potential of −70 mV (0 min). The TREK-1c current gradually increased and reached a plateau at 5 min. The application of 3 mM bupivacaine nearly completely inhibited the current. (B) The current-voltage relationship of the TREK-1c current showed outward rectification. (C and D) Inhibition of the run-up by NEM and brefeldin-A. Immediately after the first recording (gray bar), 1 mM NEM (C) or 5 µg/ml brefeldin-A (D) was added to Tyrode solution. Whole-cell conductance was normalized to that of 0 min. (* p < 0.05, the Student's t-test, n = 7). (E) Facilitation of the run-up by pitstop2. Pitstop2 (30 µM) was added to Tyrode solution immediately after whole-cell access (gray bar; * p < 0.05, n = 6).
NEM and pitstop2 changed the localization of mCherry-TREK-1c proteins
To histochemically confirm that NEM and pitstop2 affect intracellular transport of TREK-1c channels, we attempted to immunostain the channel in TR-1 cells using an anti-TREK-1 antibody (ab90855, Abcam, Cambridge, UK) and fluorescence-labeled secondary antibody. Only faint immunoreactivity was observed around the nucleus, and that at the plasma membrane was below the detectable level (data not shown). Although we used other antibodies (ab56009 and ab83932, Abcam; T6448, Sigma; NB110, Novus Biologicals), immunoreactivity levels were similar or lower. Detection of the channel appeared to be difficult with immunostaining.
To enable the visual identification of the TREK-1c channel, we fused cDNA for the TREK-1c channel with that for a red fluorescent protein, mCherry, prepared lentiviral vectors that express mCherry-TREK-1c, and then established a 293T cell line stably expressing mCherry-TREK-1c proteins (MT-1). We first confirmed the run-up of currents through mCherry-TREK-1c channels and their suppression by NEM in MT-1 cells, indicating that the N-terminal fusion of the mCherry protein did not interfere with the run-up (). We then analyzed the localization of mCherry-TREK-1c channels with red fluorescence using a confocal microscope. A single plane image showed that most mCherry-TREK-1 proteins were located intracellularly, as reported previously.Citation20,22 However, red fluorescence was also detectable at the plasma membrane in MT-1 cells (). In the statistical analysis, we categorized 100 MT-1 cells into surface expression-positive and -negative cells. Surface fluorescence was observed in 20% of cells (). We then examined the effects of NEM on the localization of mCherry-TREK-1 proteins. In MT-1 cells treated with medium containing 1 mM NEM for 3 min, fluorescence was hardly detectable at the plasma membrane () and the percentage of surface fluorescence-positive cells was significantly reduced (). Conversely, in MT-1 cells treated with medium containing 30 µM pitstop2 for 10 min, fluorescence at the plasma membrane was more prominent () and the percentage of surface fluorescence-positive cells was significantly higher () than that in control cells.
Figure 2. NEM and pitstop2 changed the localization of mCherry-TREK-1c proteins. (A and B) Inhibition of the run-up by NEM in MT-1 cells. Immediately after whole-cell access from a MT-1 cell, the TREK-1c current was evoked with a step pulse from −70 to 0 mV (0 min). Although current increased from 1 to 5 min in the control MT-1 cell, no increase was observed in the NEM-treated cell. The difference in conductance was significant (* p < 0.05, the Student's t-test, n = 5). (C) Confocal microscopic image of MT-1 cells. mCherry fluorescence was mostly located in the cytoplasm. Only weak fluorescence was observed at the plasma membrane (arrowheads). (D) NEM-treated MT-1 cells. Red fluorescence at the plasma membrane was almost diminished. (E) Pitstop2-treated MT-1 cells. Fluorescence at the plasma membrane was more prominent. (F and G) MT-1 cells were manually categorized into surface fluorescence-positive cells (arrowheads in C) and -negative cells (D). Values represent the mean and SEM of the percentage of cells categorized into surface-positive cells from 4 independent experiments, and was analyzed with the χ2-test (p < 0.001). The surface fluorescence was higher in pitstop2 treated cell as compared with NEM treatment. (H) Inhibitory effect of NEM examined with biotinylation. Cell surface proteins of MT-1 cells, which were treated with NEM (1 mM) for 3 min, were biotinylated and precipitated with streptavidin beads after solubilization. Biotinylated and Streptavidin-precipitated (i.e., surface-located, indicated as Av-ppt) mCherry-TREK proteins were analyzed with immunoblotting with anti-mCherry antibody. Immunoblots of loading control and non-biotinylated control are shown as Input and Non-biotinylated, respectively. The arrowhead indicates the position of mCherry-TREK. (I) Densitometric analysis of biotinylated mCherry-TREK. NEM-treatment significantly decreased biotinylated mCherry-TREK channel. Ordinate indicates arbitrary units of the densitometer (* p < 0.05, Student's t-test, n = 5). (J) Rapid internalization of mCherry-TREK channel. Surface located proteins were biotinylated at 4°C, and cells were incubated at 37°C for 0, 1, and 3 min to allow internalization. After cleavage of the link in the biotinylation reagent with GSH, GSH-resistant (i.e., internalized) mCherry-TREK was analyzed with immunoblotting. Only one min incubation significantly increased the internalized channel (* p < 0.05, Student's t-test, n = 5). The Input indicates loading control.
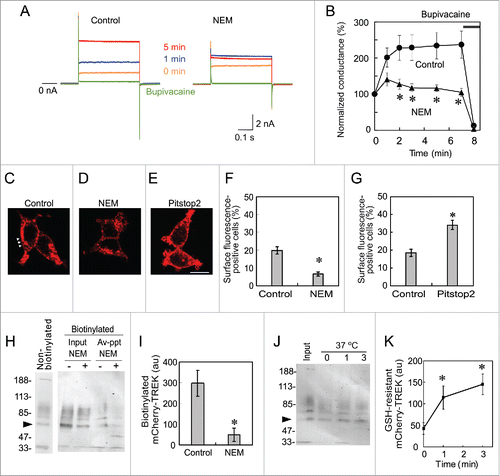
These electrophysiological and morphological data suggest rapid transport and internalization of the channel. To confirm the rapid transport to the plasma membrane, we next examined surface localization by biotinylation. MT-1 cells were first treated with a membrane transport inhibitor, NEM, for 3 min. Then proteins located at the plasma membrane were biotinylated with EZ-Link Sulfo-NHS-SS-Biotin, which is a water-soluble N-hydroxysulfosuccinimide ester biotinylation reagent whose linker includes a cleavable disulfide bond (see later). Biotinylated proteins were precipitated with streptavidin magnetic beads and analyzed by immunoblotting using anti-mCherry antibody. As expected, NEM treatment reduced biotinylated mCherry-TREK channel (), indicating the reduced surface localization of mCherry-TREK-1 channel by NEM treatment.
We next confirmed the rapid internalization of the channel by cleaving the linker of the biotinylation reagent. Once a protein is biotinylated with EZ-Link Sulfo-NHS-SS-Biotin, the link between protein and biotin can be cleaved with a reducing agent, glutathione (GSH). But if the biotinylated proteins are internalized, they will be resistant to extracellular GSH: GSH-resistance namely indicates the internalization. MT-1 cells were initially treated with biotinylation reagent at 4°C, to prevent internalization, and then incubated at 37°C for 1 or 3 min to allow internalization. Subsequently, the cells were incubated at 4°C with GSH for the cleavage of the disulfide bond, and the GSH-resistant and biotinylated TREK channels were analyzed with immunoblotting with anti-mCherry antibody (). The GSH-resistant channel was significantly increased with the incubation at 37°C for 1 and 3 min, indicating rapid internalization of the channel.
Involvement of Mtap2 in the TREK run-up
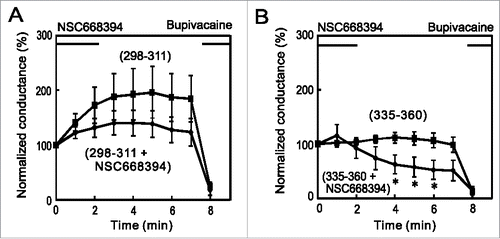
K2P channels have binding partners that influence channel function as well as trafficking in the channel to the plasma membrane. AKAP150Citation27 and Mtap2Citation19 have been identified to bind to TREK-1 channels. The binding of AKAP150 to the regulatory domain in the C-terminus is known to stimulate channel activity,Citation27 while that of Mtap2 promotes the surface expression of TREK-1 channels.Citation19 To elucidate the mechanism underlying the run-up in more detail, we synthesized the peptides, TREK298–311 and TREK335–360, which correspond to the interaction sites with AKAP150 and Mtap2, respectively. We then added these peptides to the pipette solution, and examined their effects on the run-up. Although the addition of TREK298–311 slightly reduced the run-up, the addition of TREK335–360 significantly reduced it (), suggesting that the interaction with Mtap2 (i.e., increased intracellular transport) is more important for the run-up.
Figure 3. Inhibition of the run-up by a C-terminus peptide that interacts with Mtap2. (A) Whole cell voltage-clamp recordings were made from TR-1 cells with an internal solution containing 10 μM TREK298–311 or TREK335–360 peptide, which corresponds to the site for the interaction with AKAP150 or Mtap2, respectively. No significant differences were observed in initial conductance among the 3 groups. Whereas TREK-298–311 slightly decreased the run-up, TREK-335–360 significantly inhibited it: conductance was significantly lower than that in control cells 5 and 7 min after whole-cell access (* p < 0.05, ANOVA followed by the Student's t-test, n = 6). (B) Co-immunoprecipitation of Mtap2 with mCherry-TREK. Lysate of MT-1 cells was immnunoprecipitated with anti-mCherry antibody and precipitate was analyzed with Mtap2 antibody. The arrowhead indicates the position of Mtap2. (C) Inhibition by TREK335–360 peptide. Citrate-treated and neutralized lysate was allowed to reassemble and immunoprecipitated with anti-mCherry antibody in the presence or absence of TREK335–360 peptide. Precipitate was analyzed with immunoblotting with anti-Mtap2 antibody. Addition of the peptide inhibited the co-immunoprecipitation.
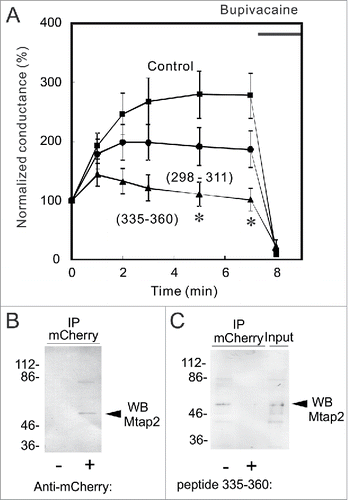
To confirm the interaction between TREK-1c and Mtap2 biochemically, we tested whether anti-mCherry antibody co-immunoprecipitates Mtap2, or not. Proteins in solubilized lysate of MT-1 cells were immunoprecipitated with anti-mCherry antibody and the precipitate was analyzed with anti-Mtap2 antibody (). Immunoblot analysis showed the co-immunoprecipitation of Mtap2 with anti-mCherry antibody, indicating the interaction between them.
We next examined the effect of TREK335–360 peptide on this co-immunoprecipitation. Initially, we added the peptide to the lysate and tried to detect the inhibition of co-immunoprecipitation of Mtap2, but failed to detect it (data not shown). This failure might be because the interaction between TREK-1c and Mtap2 is tight and hardly dissociate in cell-free lysate. Therefore, to dissociate the interaction, MT-1 cell lysate was treated with citric acid,Citation28 and the acid-treated lysate was neutralized with 1 M Tris. Proteins in the lysate were then allowed to reassemble in the presence or absence of the peptide, and analyzed with co-immunoprecipitation (). Whereas, in the absence of the peptide, mCherry-TREK successfully reassembled and co-immunoprecipitated Mtap2, the addition of TREK335–360 peptide inhibited the co-immunoprecipitation of Mtap2 with anti-mCherry antibody, indicating that the peptide inhibited the interaction between Mtap2 and C-terminal region of TREK-1c.
Involvement of ezrin in the TREK run-up
A previous study reported that TREK-1 channels and ezrin are co-localized and that the expression of TREK-1 induces the formation of actin- and ezrin-rich membrane protrusions.Citation22 These findings suggest that the interaction between TREK-1 and actin through ezrin is involved in the translocation of TREK-1 channels and the current run-up. Therefore, we applied the ezrin inhibitor, NSC668394, which inhibits ezrin phosphorylation at T567 and, thus, the binding of ezrin to F-actin,Citation29 immediately after the first recording from TR-1 cells. Although, IC50 of NSC668394 was 8.1 µM, we used 100 µM to obtain a quick response. It is reported that NSC668394 had no side effect on other kinases at this concentration.Citation29 NSC668394 significantly suppressed the run-up (), indicating the involvement of ezrin in the current run-up. Since NSC668394 was initially dissolved in 1/1000 volume of dimethylsulfoxide and then diluted to Tyrode solution before experiments, we tested the effect of the equivalent concentration of dimethylsulfoxide and found no effect on TREK-1c current (data not shown).
Figure 4. Involvement of ezrin in the surface localization of TREK-1c channels. (A) Inhibition of the run-up by an ezrin inhibitor. TREK-1c conductance gradually increased in control TR-1 cells, but the addition of the ezrin inhibitor, NSC668394 (100 µM), to Tyrode solution significantly inhibited the run-up (*p < 0.05, the Student's t-test, n = 8). (B) The percentage of cells in which mCherry-TREK-1c proteins were localized at the plasma membrane was significantly decreased by the NSC668394 treatment (p < 0.05, the χ2-test, 100 cells from 3 independent experiments). (C) The co-localization of ezrin with mCherry-TREK-1c proteins at the plasma membrane and dissociation by NSC668394. MT-1 cells were immunostained with an anti-ezrin antibody. Anti-ezrin immunoreactivity was co-localized with mCherry-TREK at the plasma membrane in control cells. In contrast, in NSC668394-treated cells, mCherry-TREK fluorescence was dissociated from the anti-ezrin antibody and plasma membrane. (D) Involvement of ezrin in the association between TREK and actin. MT-1 cells were incubated with 100 µM NSC668394 or 0.1% DMSO for 5 min. Solubilized lysates, which were immunoprecipitated with an anti-mCherry antibody, were then immunoblotted with an anti-actin antibody. The addition of NSC668394 decreased actin precipitation, suggesting the involvement of ezrin in the association of TREK-1c with actin fibers. Arrow and arrowheads indicate the position of actin and IgG, respectively.
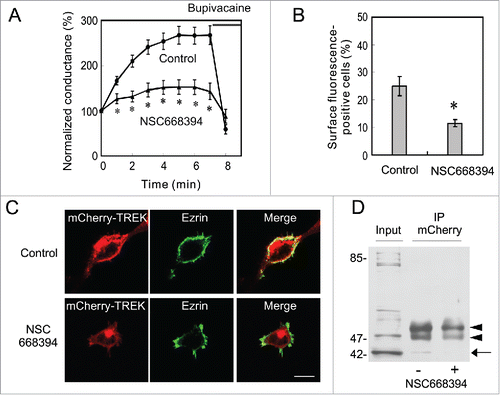
We then examined the localization of mCherry-TREK-1c and ezrin, and the effects of NSC668394 on the surface expression of TREK-1c in MT-1 cells. Single plane confocal images showed that ezrin was localized at the plasma membrane and overlapped with mCherry-TREK-1c at the cell surface (). The treatment of MT-1 cells with NSC668394 diminished the surface expression of TREK-1c and overlapping: anti-ezrin immunoreactivity was observed at the plasma membrane, whereas mCherry-TREK fluorescence was negligible. The percentage of MT-1 cells with the surface localization of mCherry-TREK-1c was significantly reduced in NSC668394-treated cells (), suggesting the important role of ezrin in the surface localization of TREK-1c channels.
Since ezrin interacts with actin fibers,Citation7 we tested the effects of NSC668394 on the interaction between TREK-1c and actin. MT-1 cells were incubated with NSC668394 or 0.1% DMSO (negative control), and cell lysates were immunoprecipitated with the anti-mCherry antibody and analyzed with immunoblotting using the anti-actin antibody (). The anti-actin antibody successfully detected a band at 42 kDa in control lysates, whereas only a faint band was observed in NSC668394-treated cells, suggesting the interaction of TREK-1c channels with actin fibers through ezrin.
Additive effects of Mtap2 and ezrin in the regulation of TREK-1c channels
The results presented so far show that Mtap2 and ezrin are involved in the run-up of TREK-1c currents and raise the question of whether they enhance the run-up interdependently or additively. If Mtap2 and ezrin interdependently enhance the run-up in the same pathway, the co-application of inhibitors may not suppress the run-up further. On the other hand, if these proteins additively enhance the run-up in separate pathways, the co-application of inhibitors may result in the further suppression of the run-up. To test these possibilities, we perfused TR-1 cells with Tyrode solution containing NSC668394 after the first recording with pipette solution containing the TREK335–360 peptide. The TREK-1c currents showed a further decrease (), thereby demonstrating the additive effect in separate pathways. The co-application of NSC668394 and TREK298–311, corresponding to the binding site with AKAP150, served as a negative control, excluding the possibility of the negative effects of the co-application ().
Figure 5. Additive effects of ezrin and Mtap2 on the run-up of the TREK-1c current. (A) A whole-cell patch-clamp was made from TR-1 cells with internal solution containing 10 µM TREK-298–311, which corresponds to the binding site with AKAP150. NSC668394 (100 µM) was applied immediately after the first recording. The effects of the co-application of NSC668394 and TREK298–311 were similar to those of NSC668394 alone (compare with ). (B) The co-application of NSC668394 with TREK-335–360, which corresponds to the binding site with Mtap2, diminished the TREK-1c current. Conductance was significantly lower than that of TREK-335–360 alone (* p < 0.05, the Student's t-test, n = 7). This result suggests the additive involvement of Mtap2 and ezrin in the run-up.
Discussion
TREK-1 currents in cells expressing wild-type channels always increased in the few minutes following whole-cell access. The present study clearly showed the involvement of intracellular transport in the run-up of the TREK-1c current. 1) The NEM and brefeldin-A treatment inhibited the run-up and decreased the surface expression of mCherry-TREK. 2) The pitstop2 treatment facilitated the run-up and increased surface expression. 3) The biotinylation experiments confirmed the rapid transport of the channel to the plasma membrane and internalization. 4) The addition of TREK335–360, the peptide corresponding to the interaction site with Mtap2, to the pipette solution inhibited the run-up. Furthermore, we used an ezrin inhibitor and showed that ezrin affected the run-up in a different manner to Mtap2. These results suggest 2 features of the transport of TREK-1 channel. The transport to the plasma membrane and internalization were rapid, which were accomplished within a few minutes. Trafficking of cystic fibrosis transmembrane conductance regulator was reported to be similarly rapid.Citation30 On the other hand, when pitstop2 inhibits endocytosis to facilitate the run-up, the maximal value of conductance was similar to that of the control. This is probably because the number of vesicles, which are readily fusible to the plasma membrane, is limited.
Proteins of the ezrin, radixin, and moesin (ERM) family act as dynamic linkers between the actin cytoskeleton and plasma membrane.Citation31 When the N- and C-terminal domains interact intramolecularly to mask an F-actin binding site in the C-terminus, ERM proteins are dormant and localized in the cytoplasm. The phosphorylation of highly conserved Thr residues and the binding of phosphatidylinositol 4,5-bisphosphate change ERM proteins to their active forms, which interact with plasma membrane proteins and F-actin. The N-terminal domain has been shown to interact with membrane proteins directly or indirectly through scaffolding proteins such as EBP50.Citation31 Lauritzen et al. reported that TREK-1 channels and ezrin are co-localized and also that the expression of TREK-1 induces the formation of actin- and ezrin-rich membrane protrusions.Citation22 Our results showing that the ezrin inhibitor decreased the run-up of the TREK-1c current (), the surface localization of the channel (), and a band corresponding to actin () suggest that TREK-1 channels interact with ezrin to be stabilized in the plasma membrane. Mtap2 interacts with the C-terminal domain of TREK-1 (Glu335-Gln360) and also binds to microtubules, thereby increasing the transport and cell surface expression of the channel.Citation19 The present results showing that inhibitors of Mtap2 and ezrin depressed TREK-1c currents in an additive manner suggest that Mtap2 and ezrin promote the surface expression of the channel by enhancing intracellular transport and stabilization, respectively.
Previous study reported the run-up of TREK current in excised patches.Citation32 Because endocytosisCitation33 and exocytosisCitation34 were also observed in excised patches from INS-1 and chromaffin cells, it is likely that excised patches have the machinery for transport so that the TREK channels are potentially transported to the excised patch membrane.
Although we used 293T cell lines in the present study, the same results are expected in neurons and other excitable cells. The TREK-1 current also showed the run-up in adrenocortical cells,Citation23 which secrete glucocorticoids. In addition, 2-pore-domain K+ channels are known to be upregulated to maintain appropriate excitability in central neurons.Citation35 Previous studies demonstrated that TREK-1, AKAP150, and Mtap2 are present in the postsynaptic dense bodies of hippocampal neurons.Citation19,27 The molecular mechanisms underlying the run-up may play an important role in the regulation of endocrine secretion and neuronal excitability. Segal-Hayoun et al. have shown the membrane potential dependent regulation of TREK-1 current in Xenopus oocytes.Citation36 We similarly have found that surface localization of TREK channel was changed depending on membrane potential (data not shown). This dependency is probably relevant to the physiologic role of channel trafficking.
Materials and methods
Plasmid construction
The cDNA of TREK-1c was generously donated by Dr. Wischmeyer (University of Würzburg). We prepared a lentiviral vector that expresses GFP and TREK-1c channelsCitation24 and, using this viral vector, established the 293T cell line, TR-1, which stably expresses these proteins, with limited dilutions. The mCherry-TREK-1c fusion gene was constructed with PCRs and cloned to the lentiviral self-inactivating expression plasmid CS-β-actinP. CS-β-actinP prepared from CS-CDF-CG-PRE (provided by Dr. Miyoshi, RIKEN) contained a chick β-actin promoter instead of a CMV promoter. We prepared a lentiviral vector expressing mCherry-TREK-1c and established the stably expressing cell line, MT-1, in the same manner. All experiments were approved by the Committee of Gene Recombination Experiments of Kansai Medical University.
Patch-clamp recordings
TR-1 and MT-1 cells were maintained in DMEM containing 10% FBS and penicillin/streptomycin under a humidifying atmosphere containing 5% CO2 at 37°C. The coverslips on which cells were grown were transferred to a recording chamber on the stage of an inverted microscope (Olympus IX70, Tokyo, Japan). Whole-cell currents were recorded in Tyrode solution using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) at 25°C. Tyrode solution contained (in mM): NaCl 140, KCl 5.4, NaH2PO4 0.33, CaCl2 2, MgCl2 1, HEPES 5, and glucose 5.5 (pH 7.4 adjusted with NaOH). Patch pipettes pulled from borosilicate glass (Narishige, Tokyo, Japan) were filled with an internal solution containing (in mM): K-aspartate 66, KCl 71.5, KH2PO4 1, EGTA 5, Hepes 5, and K2ATP 3 (pH 7.4 adjusted with KOH). Records were digitized at 10 kHz, and low-pass filtered at 2 kHz. Step pulses of 400 ms from −100 to 40 mV were applied in 10-mV increments from a holding potential of −70 mV. Slope conductance was calculated based on the current–voltage relationship from −70 to 0 mV and was normalized at each time point with that of the first estimation. Data are shown as the mean ± SE. N-methylmaleimide (NEM; 1 mM), brefeldin-A (5 µg/ml), and pitstop2 (30 µM) were added to Tyrode solution. The partial peptides corresponding to the binding sites of TREK-1 to AKAP150 (TREK298–311) and Mtap2 (TREK335–360) were chemically synthesized (Peptide2.0, Chantilly, VA) and added to the internal solution at a concentration of 10 μM. The ezrin inhibitor, NSC668394 (Merk Millipore, Darmstadt, Germany) was added to Tyrode solution (100 μM).
Confocal microscopic analysis of MT-1 cells
MT-1 cells cultivated in a 35-mm dish were treated at 37°C with 1 mM NEM for 3 min, 30 µM pitstop2 for 10 min, or 100 µM NSC668394 for 3 min and then fixed with 4% paraformaldehyde at room temperature for 30 min. The cells were washed with PBS, and single plane confocal images were taken with FV 300 (Olympus, Tokyo, Japan) using Texas Red filter sets. At the beginning of the experiment, parameters, i.e., laser intensity, gain, and the offset value, were adjusted and maintained in a series of experiments. For statistical analysis, percentage of surface mCherry-TREK fluorescence-positive cells was estimated: one hundred cells, without signs of degeneration, were manually selected and classified into surface fluorescence-positive (e.g., arrowheads in ) and -negative cells ().
Biotinylation of surface proteins
For the analysis of inhibition of transport to the plasma membrane, MT-1 cells were incubated with NEM (1 mM) for 3 min at 37°C. Then cell surface proteins were biotinylated with Sulfo-NHS-SS-Biotin (Thermo Fisher scientific, Waltham, MA, dissolved in PBS, pH 8.2) for 2 h at 4°C and washed with PBS (pH8.2) 3 times.Citation30 Then MT-1 cells were solubilized with lysis buffer containing 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.1% SDS, 1% Nonidet P-40, 0.5% sodium cholate, and protease inhibitor cocktail (08714, Nacalai Tesque, Kyoto, Japan) on ice for 15 min. Cell suspensions were centrifuged at 10,000 x g for 15 min, and the supernatants were collected as cell lysate. The biotinylated proteins were recovered with streptavidin magnetic beads according to the instruction manual (Pierce, Rockford, IL, USA) and subjected to immunoblot analysis with anti-mCherry antibody. The immunoreaction was analyzed with a GS-900 densitometer (Bio-Rad Laboratories, Hercules, CA).
For the analysis of internalization of the channel, cell surface proteins were first biotinylated with Sulfo-NHS-SS-Biotin, and MT-1 cells were incubated at 37°C for 1 or 3 min to allow internalization. The linker of the biotinylated residue, which remained at the plasma membrane, was cleaved by reducing agent (50 mM GSH, 4°C, 15 min x 6 times).Citation30 After washing 3 times with PBS (pH 8.2), cells were lysed as described above. Then the GSH-resistant, i.e., internalized, proteins were recovered with streptavidin magnetic beads and analyzed with immunoblotting with anti-mCherry antibody.
Inhibition of co-immunoprecipitation of Mtap2 with TREK335–360 peptide
To dissociate protein bindings, we added 15 µl of 0.1 M citrate (pH 2.1) to the lysates (250 µl) from MT-1 cell and incubated at 70°C for 10 min,Citation28 and subsequently neutralized it with 50 µl of 1 M Tris. Then to allow the reassembly, the neutralized lysate was incubated in the presence or absence of peptide TREK335–360 (10 µM), together with anti-mCherry antibody and Dynabeads-proteinG for immunoprecipitation for 2 hr at 4°C. Co-imunoprecipitated proteins were analyzed with immunoblot with anti-Mtap2 (1:1000; 17490–1-AP, Proteintech,Rosemont, IL).
Immunoblotting analysis
Prior to solubilization, some MT-1 cells were treated with NSC668394 for 5 min. In immunoprecipitation, cell lysates were incubated with the anti-mCherry antibody and Dynabeads protein G (Thermo Fisher Scientific, Waltham, MA). The immunoprecipitates that bound to the beads were collected and washed 3 times with washing buffer, and the proteins were eluted with NuPAGELDS Sample buffer and reducing agent according to the instruction manual (Thermo Fisher Scientific). Samples were analyzed by SDS-PAGE (7.5%) under reducing conditions and transferred to a PVDF membrane (Merk Millipore). The membrane was blocked with blocking one (Nacalai Tesque, Kyoto, Japan) and incubated with anti-actin (1:400) antibody diluted in signal enhancer Hikari solution A (Nacalai Tesque) and then with a secondary antibody conjugated with alkaline phosphatase (Promega, Fitchburg, WI) diluted in Hikari solutin B (Nacalai Tesque). Regarding the colorimetric detection of alkaline phosphatase activity, BCIP (5-bromo-4-chloro-3-indolyl-phosphate) was used in conjunction with NBT (nitro blue tetrazolium; Promega).
Immunostaining
Antibodies against mCherry (ab183628) and actin (ab3280) were purchased from Abcam (Cambridge, UK). Antibody against ezrin (Ezrin/p81/80K/Cytovillin Ab-1(3C12)) was purchased from Thermo Fisher Scientific (Waltham, MA). MT-1 cells were fixed with 4% paraformaldehyde, permeabilized with PBS containing 0.3% Triton X-100 and 0.3% BSA, and reacted with the first antibodies. The immunoreaction was visualized using a secondary antibody conjugated with Alexa Fluor 488 (A11029, Invitrogen, Carlsbad, CA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by KAKENHI (25460323) from the JSPS, the SICP from the JST, the Heiwa Nakajima Foundation, the Ryobi Memorial Foundation, Wesco Scientific Promotion Foundation (to MO), the Mishima Memorial Foundation, the Rotary Yoneyama Memorial Foundation (to NA), the Fujii Memorial Foundation (to AJ), and KAKENHI (24790226, to MH).
Related Research Data
References
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M. Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 1996; 15(24):6854-6862; PMID:9003761
- Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, Fosset M, Lazdunski M. A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J 1998; 17(12):3297-3308; PMID:9628867; https://doi.org/10.1093/emboj/17.12.3297
- Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev 2005; 57(4):527-540; PMID:16382106; https://doi.org/10.1124/pr.57.4.12
- Goldstein SA, Bockenhauer D, O'Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci 2001; 2(3):175-184; PMID:11256078; https://doi.org/10.1038/35058574
- Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA. CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci 2001; 21(19):7491-7505; PMID:11567039
- Terrenoire C, Lauritzen I, Lesage F, Romey G, Lazdunski M. A TREK-1-like potassium channel in atrial cells inhibited by β-adrenergic stimulation and activated by volatile anesthetics. Circ Res 2001; 89(4):336-342; PMID:11509450; https://doi.org/10.1161/hh1601.094979
- Turunen O, Wahlström T, Vaheri A. Ezrin has a COOH-Terminal actin-binding site that is conserved in the ezrin protein family. J Cell Biol 1994; 126(6):1445-1453; PMID:8089177; https://doi.org/10.1083/jcb.126.6.1445
- Xian Tao Li, Dyachenko V, Zuzarte M, Putzke C, Preisig-Müller R, Isenberg G, Daut J. The stretch-activated potassium channel TREK-1 in rat cardiac ventricular muscle. Cardiovasc Res 2006; 69(1):86-97; PMID:16248991; https://doi.org/10.1016/j.cardiores.2005.08.018
- Honoré E, Maingret F, Lazdunski M, Patel AJ. An intracellular proton sensor commands lipid- and mechano-gating of the K+ channel TREK-1. EMBO J 2002; 21(12):2968-2976; PMID:12065410; https://doi.org/10.1093/emboj/cdf288
- Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol 2000; 279(5):F793-801; PMID:11053038
- Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honoré E. TREK-1 is a heat-activated background K + channel. EMBO J 2000; 19(11):2483-2491; PMID:10835347; https://doi.org/10.1093/emboj/19.11.2483
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E. Lysophospholipids open the two-pore domain mechano-gated K+ channels TREK-1 and TRAAK. J Biol Chem 2000; 275(14):10128-10133; PMID:10744694; https://doi.org/10.1074/jbc.275.14.10128
- Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci 1999; 2(5):422-426; PMID:10321245; https://doi.org/10.1038/8084
- Murbartián J, Lei Q, Sando JJ, Bayliss DA. Sequential phosphorylation mediates receptor- and kinase-induced inhibition of TREK-1 background potassium channels. J Biol Chem 2005; 280(34):30175-30184; PMID:16006563; https://doi.org/10.1074/jbc.M503862200
- Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, Lang-Lazdunski L, Widmann C, Zanzouri M, Romey G, et al. TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J 2004; 23(13):2684-2695; PMID:15175651; https://doi.org/10.1038/sj.emboj.7600234
- Schulz DJ, Goaillard JM, Marder EE. Quantitative expression profiling of identified neurons reveals cell-specific constraints on highly variable levels of gene expression. Pro Natl Acad Sci USA 2007; 104(32):13187-13191; PMID:17652510; https://doi.org/10.1073/pnas.0705827104
- Okada M, Kano M, Matsuda H. The degradation of the inwardly rectifying potassium channel, Kir2.1, depends on the expression level: Examination with fluorescent proteins. Brain Res 2013; 1528:8-19; PMID:23850646; https://doi.org/10.1016/j.brainres.2013.07.008
- Ma D, Jan LY. ER transport signals and trafficking of potassium channels and receptors. Curr opin Neurobiol 2002; 12(3):287-292; PMID:12049935; https://doi.org/10.1016/S0959-4388(02)00319-7
- Sandoz G, Tardy MP, Thümmler S, Feliciangeli S, Lazdunski M, Lesage F. Mtap2 is a constituent of the protein network that regulates twik-related K+ channel expression and trafficking. J Neurosci 2008; 28(34):8545-8552; PMID:18716213; https://doi.org/10.1523/JNEUROSCI.1962-08.2008
- Kim E, Hwang EM, Yarishkin O, Yoo JC, Kim D, Park N, Cho M, Lee YS, Sun CH, Yi GS, et al. Enhancement of TREK1 channel surface expression by protein-protein interaction with β –COP. Biochem and Biophys Res Commun 2010; 395(2):244-250; PMID:20362547; https://doi.org/10.1016/j.bbrc.2010.03.171
- Mazella J, Pétrault O, Lucas G, Deval E, Béraud-Dufour S, Gandin C, El-Yacoubi M, Widmann C, Guyon A, Chevet E, et al. Spadin, a sortilin-derived peptide, targeting rodent TREK-1 channels: A new concept in the antidepressant drug design. PLoS Biol 2010; 8(4):e1000355; PMID:20405001; https://doi.org/10.1371/journal.pbio.1000355
- Lauritzen I, Chemin J, Honoré E, Jodar M, Guy N, Lazdunski M, Jane Patel A. Cross-talk between the mechano-gated K2P channel TREK-1 and the actin cystoskeleton. EMBO Reports 2005; 6(7):642-648; PMID:15976821; https://doi.org/10.1038/sj.embor.7400449
- Enyeart JJ, Xu L, Danthi S, Enyeart JA. An ACTH- and ATP- regulated background K+ channel in adrenocortical cells is TREK-1. J Biol Chem 2002; 277(51):49186-49199; PMID:12368289; https://doi.org/10.1074/jbc.M207233200
- Okada M, Andharia N, Matsuda H. Increase in the titer of lentiviral vectors expressing potassium channels by current blockade during viral vector production. BMC Neurosci 2015; 16:30; PMID:25940378; https://doi.org/10.1186/s12868-015-0159-1
- Shin HW, Soh JS, Kim HZ, Hong J, Woo DH, Heo JY, Hwang EM, Park JY, Lee CJ. The inhibitory effects of bupivacaine, levobupivacaine, and ropivacaine on K2p (two-pore domain potassium) channel TREK-1. J Anesth 2014; 28(1):81-96; PMID:23797625; https://doi.org/10.1007/s00540-013-1661-1
- Polakova K, Russ G. Use of Brefeldin A to localize block in intracellular transport of vesicular stomatitis virus G protein on interferon-treated cells. Arch Virol 1992; 124(1–2):171-179; PMID:1373940; https://doi.org/10.1007/BF01314635
- Sandoz G, Thümmler S, Duprat F, Feliciangeli S, Vinh J, Escoubas P, Guy N, Lazdunski M, Lesage F. AKAP150, a switch to convert mechano-, pH- and arachidonic acid-sensitive TREK K+ channels into open leak channels. EMBO J 2006; 25(24):5864-5872; PMID:17110924; https://doi.org/10.1038/sj.emboj.7601437
- Remily-Wood E, Dirscherl H, Koomen JM. Acid hydrolysis of proteins in matrix assisted laser desorption ionization martices. J Am Soc Mass Spectrom 2009; 20(11):2106-2115; PMID:19679491; https://doi.org/10.1016/j.jasms.2009.07.007
- Bulut G, Hong SH, Chen K, Beauchamp EM, Rahim S, Kosturko GW, Glasgow E, Dakshanamurthy S, Lee HS, Daar I, et al. Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene 2012; 31(3):269-281; PMID:21706056; https://doi.org/10.1038/onc.2011.245
- Cihil KM, Swiatecka-Urban A. The cell-based L-glutathione protection assays to study endocytosis and recycling of plasma membrane proteins. J Vis Exp 2013; 13:e50867; PMID:24378656; https://doi.org/10.3791/50867
- Bretscher A, Edwards K, Fehon RG. ERM protein and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 2002; 3(8):586-599; PMID:12154370; https://doi.org/10.1038/nrm882
- Hwang SJ, O'Kane N, Singer C, Ward SM, Sanders KM, Koh SD. Block of inhibitory junction potentials and TREK-1 channels in murine colon by Ca2+ store-active drugs. J Physiol 2008; 586(4):1169-1184; PMID:18187470; https://doi.org/10.1113/jphysiol.2007.148718
- MacDonald PE, Eliasson L, Rorsman P. Calcium increases endocytotic vesicle size and accelerates membrane fission in insulin-secreting INS-1 cells. J Cell Sci 2005; 118(24):5911-5920; PMID:16317049; https://doi.org/10.1242/jcs.02685
- Derrick G, Alvarez de Toledo G, Lindau M. Exocytosis of single chromatin granules in cell-free inside-out membrane patches. Nat Cell Biol 2003; 5(4):358-362; PMID:12652310; https://doi.org/10.1038/ncb956
- Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M. Adaptive regulation of neuronal excitability by a voltage- independent potassium conductance. Nature 2001; 409(6816):88-92; PMID:11343119; https://doi.org/10.1038/35051086
- Segal-Hayoun Y, Cohen A, Zilberberg N. Molecular mechanisms underlying membrane-potential-mediated regulation of neuronal K2P2.1 channels. Mol Cell Neurosci 2010; 43(1):117-126; PMID:19837167; https://doi.org/10.1016/j.mcm.2009.10.002