
Bartter syndrome is caused by mutations in several salt-handling channels in the kidney, but the type II variety is particularly severe. The disease usually presents in utero with polyhydramnios, increasing the risk of premature birth.Citation1 Infants with type II Bartter syndrome develop hypercalciuria, nephrocalcinosis, and osteopenia. Fever, emesis, dehydration, and diarrhea are symptoms associated with the disease, which is secondary to enhanced prostaglandin E2 activity. Ultimately, Bartter Syndrome is characterized by polyuria, hyponatremia, hypokalemia, metabolic alkalosis, and failure to thrive, which can progress to end-stage renal failure. There is no cure for this disease.
Type II Bartter syndrome arises from mutations in the gene encoding the renal outer medullary potassium channel, “ROMK," which is a member of the inwardly-rectifying family of potassium channels. ROMK is expressed at the apical surface of epithelial cells in the distal nephron, including the thick ascending limb of the loop of Henle and the cortical collecting duct. There are three ROMK isoforms, ROMK1-3, each with different N-termini generated by alternative splicing.Citation2 The isoforms exhibit similar biophysical properties but are expressed in different segments along the nephron. Mutations in the common sequence of any of the isoforms lead to type II Bartter syndrome, and among the ∼60 disease-causing mutations, N-terminal nonsense or frameshift mutations have obvious deleterious consequences. Other mutations can be grouped into two categories: point mutations which disrupt gating, ion selectivity, or ligand binding, and those which disrupt trafficking and reduce ROMK at the plasma membrane. Previous studies demonstrated that some of these mutations impede trafficking when expressed in Xenopus oocytes or HEK293 cells.Citation3,Citation4
Among the disease-causing mutations that affect protein trafficking, four (A198T, R212P, H270Y, and Y314C) are clustered in a β-sheet-rich immunoglobulin-like domain in the C-terminal region of ROMK that resides in the cytosol. Because β-sheet-rich regions are more difficult to fold—and because numerous protein conformational diseases arise from the aggregation of β-sheet-rich proteins5—we hypothesized that these mutant proteins would fail to achieve their native conformations. Instead, they might be trapped in the endoplasmic reticulum (ER) and targeted for destruction by the ER-associated degradation pathway, or “ERAD.” Nearly 70 human diseases are linked to ERAD, which clears the ER of misfolded, aggregation-prone proteins. Many of these diseases arise from defects in the folding of ion channels and transporters.Citation6
To test our hypothesis, we developed a new ROMK expression system in yeast cells that lack endogenous potassium channels. To survive on low potassium, the yeast require an active potassium channel at the plasma membrane. We also introduced two amino acid substitutions into the gene encoding ROMK to improve ER exit and prevent channel inhibition at low pH, since the yeast cytoplasm is acidic. As hoped, yeast expressing the modified version of ROMK grew on low potassium media. In contrast, yeast expressing ROMK containing the A198T, R212P, H270Y, or Y314C mutations in the β-sheet-rich domain were inviable.Citation7
Based on the ease with which the contributions of factors required for ERAD can be investigated in yeast, we then examined the stability of ROMK as well as the four type II Bartter syndrome mutants in strains lacking several ERAD-requiring factors, including an ATPase that extracts substrates from the ER and an Hsp70 that selects misfolded proteins. Both factors were required for degradation of the Bartter mutant proteins. Moreover, we discovered that ROMK containing the disease-causing mutations was significantly less stable than ROMK lacking these mutations. We also found that proteasome activity was required to degrade the mutant proteins, and they resided exclusively in the ER prior to degradation, consistent with disposal via the ERAD pathway.
Are these disease-associated proteins also targeted for ERAD in human cells? We next expressed wild type ROMK or the A198T, R212P, H270Y, and Y314C mutants in HEK293 cells. As observed in yeast, the mutant proteins were less stable than wild type ROMK and degradation was again proteasome-dependent. Finally, to determine whether the folding defect in the mutants might be correctable, we incubated cells at low temperature. This treatment stabilized the proteins, suggesting that small molecule “correctors” can be developed to overcome folding defects associated with specific type II Bartter mutations.Citation7
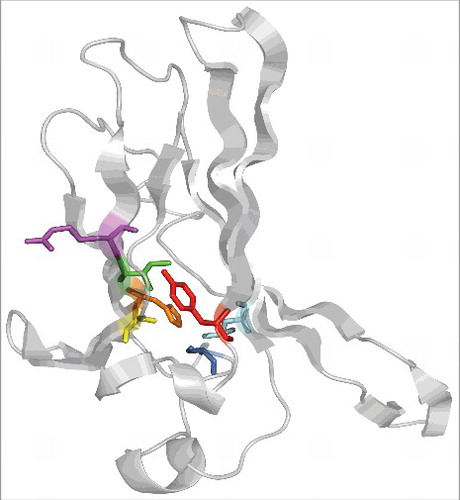
Previous work established that a residue altered in one of the mutants, Y314, stabilized the hydrophobic core of the immunoglobulin-like domain.Citation8 Indeed, a mutation at this site (Y314C) and another mutation examined in our study (A198T) alter side chains that face the hydrophobic interior of this domain (). We propose that other uncharacterized disease-causing mutations, I211S and L297S, will similarly destabilize ROMK since they also replace core-facing hydrophobic side chains with hydrophilic groups. In turn, two other mutations examined in our work, R212P and H270Y, may respectively alter main chain topology (R212P) and interfere with a hydrogen bond between the imidazole nitrogen in H270 (H270Y) and a hydroxyl in S276. Interestingly, H270 and S276 are conserved in all members of the human Kir potassium channel family to which ROMK belongs. Therefore, we propose that the S276N disease-causing mutation will also trigger ROMK degradation via ERAD.
Overall, these studies add type II Bartter syndrome to the expanding list of diseases associated with ERAD, a number that is sure to rise as genome sequence and patient databases expand.
Figure 1. The ROMK immunoglobulin-like domain with select residues mutated in Bartter syndrome are highlighted. Mutations tested in the recently published study: A198T (yellow), R212P (purple), H270Y (orange), Y314C (red). Other mutations of interest and discussed in the text: I211S (green), S276N (cyan), L297S (dark blue). Representations rendered with PyMol molecular graphics system version 1.8.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Bhat YR, Vinayaka G, Sreelakshmi K. Antenatal bartter syndrome: a review. Int J Pediatr. 2012;2012:857136.
- Welling PA. Roles and Regulation of Renal K Channels. Annu Rev Physiol. 2016;78:415-35. doi:10.1146/annurev-physiol-021115-105423. PMID:26654186.
- Peters M, Ermert S, Jeck N, Derst C, Pechmann U, Weber S, Schlingmann KP, Seyberth HW, Waldegger S, Konrad M. Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int. 2003;64:923-32. doi:10.1046/j.1523-1755.2003.00153.x. PMID:12911542.
- Fang L, Li D, Welling PA. Hypertension resistance polymorphisms in ROMK (Kir1.1) alter channel function by different mechanisms. Am J Physiol Renal Physiol. 2010;299:F1359-64. doi:10.1152/ajprenal.00257.2010. PMID:20926634.
- Beerten J, Schymkowitz J, Rousseau F. Aggregation prone regions and gatekeeping residues in protein sequences. Curr Top Med Chem. 2012;12:2470-78.
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537-76. doi:10.1152/physrev.00027.2011. PMID:22535891.
- O'Donnell BM, Mackie TD, Subramanya AR, Brodsky JL. Endoplasmic Reticulum-Associated Degradation of the Renal Potassium Channel, ROMK, Leads to Type II Bartter Syndrome. J Biol Chem. 2017; 292:12813-12827. doi:10.1074/jbc.M117.786376. PMID:28630040.
- Fallen K, Banerjee S, Sheehan J, Addison D, Lewis LM, Meiler J, Denton JS. The Kir channel immunoglobulin domain is essential for Kir1.1 (ROMK) thermodynamic stability, trafficking and gating. Channels (Austin). 2009; 3:57-68. doi:10.4161/chan.3.1.7817. PMID:19221509.