
ABSTRACT
The family of P-loop channels, which play key roles in the cell physiology, is characterized by four membrane re-entering extracellular P-loops that connect eight transmembrane helices of the pore-forming domain. The X-ray and cryo-EM structures of the open- and closed-state channels show conserved state-dependent folding despite the sequences are very diverse. In sodium, calcium, TRPV and two-pore channels, the pore-lining helices contain conserved asparagines and may or may not include π-helix bulges. Comparison of the sequence- and 3D-alignemnts suggests that the asparagines appeared in evolution as insertions that are accommodated in two ways: by π-helix bulges, which preserve most of inter-segment contacts, or by twists of the C-terminal thirds and switch of inter-segment contacts. The two possibilities should be considered in homology modeling of ion channels and in structure-based interpretations of numerous experimental data on physiology, pathophysiology, pharmacology and toxicology of the channels.
Introduction
The family of P-loop channels is characterized by tetrameric or pseudo-tetrameric structures with membrane-reentering P-loops, which harbor selectivity-filter residues. This very diverse family includes, but not limited by voltage-gated potassium, sodium, calcium channels, TRP channels, glutamate- and cyclic nucleotide-gated channels. The channels play key roles in physiology and are targeted by many toxins and medicinally important drugs. Numerous channelopathies are described for human P-loop channels.
In the last two decades structures of various P-loop channels have been obtained with the X-ray crystallography, e.g.Citation1–7 and cryoelectron microscopy, e.g.Citation8,9 Voltage-gated channels (4 × 6TM) have four voltage-sensing domains and a pore domain. The later contains four outer helices (S5s), four P-loops and four pore-lining inner helices (S6s). P-loop channels adopt substantially different conformations in the open and closed states. In 3D structures of both open and closed P-loop channels, membrane-descending P1 helices in P-loops appear the most conserved segments that may be superimposed to obtain 3D alignments. Despite impressive progress in the experimental structural biology of P-loop channels, 3D structures of many eukaryotic channels are still unavailable and homology modeling is used to elaborate structural rationale for numerous data obtained by mutational, electrophysiological and ligand-binding experiments. The homology modeling approach is generally validated by the fact that overall folding of P-loop channels is conserved despite their sequences, functional properties, and structural peculiarities of certain regions may be highly diverse. For example, potassium and sodium channels have substantially different structures of the selectivity filters and dimensions of subunit/repeat interfaces.Citation1,10 The latter peculiarities are important for understanding how drugs access the closed channels.
A key step in homology modeling is the sequence alignment between a template channel for which experimental 3D structure is available and a query channel to be modeled. When the first X-ray structures of potassium channels become available,Citation2,11 various sequence alignments were proposed between these channels and medically important sodium and calcium channels.Citation3,12–14 More recent 3D structures of sodium,Citation4,6,7,10 calcium,Citation9 TRPV1Citation15 and two-poreCitation16 channels opened a possibility to obtain 3D alignment of various P-loop channels, compare sequence-based and 3D-based alignments and analyze similarity and differences of these channels. Results of this comparison reveal some problems in sequence alignments that previously seemed unambiguous. In particular, functionally important and apparently conserved residues, which in the aligned sequences of homologous P-loop channels occur in matching positions, in 3D structures may have opposite orientations. For example, the finger-print motif GYG in potassium channels misaligns with apparently homologous GMG motif in the TRPV1 channel.Citation5,17 Possible structural diversity of the inner helices in P-loop channels has not yet been analyzed.
Here we compared 3D structures of different P-loop channels and found that the inner helices in some channels adopt classical alpha-helical conformations, while in other channels they may have short π-helical bulges (TRPV1, NavPaS, and two-pore channel TPC1) or 310 helices (at the C-ends of the open NavAb channel). Thus, residues that appear in matching positions of sequence alignments may have different orientations and different functionally important contacts. These facts must be taken into account in structural interpretation of mutational, electrophysiological, ligand-binding and other experiments, as well as in analyses of channelopathy-causing mutations. Our analysis further suggests phylogenetic relations of some P-loop channels.
Results and discussion
The sequence alignment of potassium and bacterial sodium channels is consistent with their 3D structures
The 3D aligned structures are shown in . Within the family of potassium channels, alpha carbons of P1 helices deviate from the reference Kv1.2 structure by ∼ 1 Å. For bacterial sodium channels respective deviations from the Kv1.2 structure are in the range of 1.5 to 3.0 Å. The 3D alignment was performed by minimizing the RMS deviations of only the matching alpha carbons in the P1 helices. However, the outer and inner helices also 3D aligned rather well (,). Numerical characteristics of 3D similarity of the inner helices are shown in , . At the N-end halves of the inner helices, which form close contacts with the P1 helices, the deviations of alpha carbons from Kv1.2 are rather small. The deviations increase above 10 Å at the C-terminal halves of the inner helices, which contain residues contributing to the activation gate; positions of these residues obviously differ between the open- and closed-state structures. Alpha carbon deviations of the open-state sodium channels NavAbCitation6 and NavMsCitation4 from the open-state Kv1.2 are rather small all along the S6 helices (). For the open-state NavCt channelCitation18 the deviations are large (), but residues in matching S6 positions of the alignment () have similar orientations and mutual disposition (). Thus, despite significant structural differences between the open and closed potassium and bacterial sodium channels, the sequence alignment matches the 3D alignment.
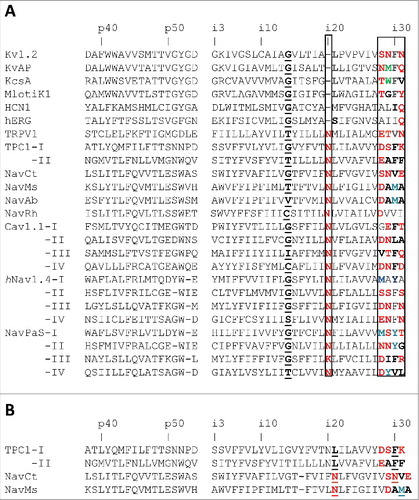
Figure 1. Sequence and 3D alignment of P-loops and inner helices in P-loop channels. A, Conventional sequence alignment. The top row shows universal labels.Citation3 P-loops of the TRPV channels are aligned as inCitation17 and in Nav1.4 and NavPaS channels they are aligned as in.Citation32 Gating-hinge residues (position i14) are underlined. Residues in position i20 and at the C-ends of the inner helices (positions i27 – i30 or i28 – i31) are boxed. Polar residues at the C-ends are red and predominantly hydrophobic residues, whose side chains may form H-bonds, are green. Note that in most of the sequences the inner-helix C-ends contain tetrads of polar-polar-hydrophobic-polar residues (motif p2hp). In the sodium, calcium, TRP and TPC channels, which have asparagines Ni20, are the p2hp motif is shifted vs. potassium channels, which have with leucines Li20. B and C, Side and extracellular views of the 3D aligned structures. The outer helices, P-loops and inner helices are green, blue and red, respectively. The 3D alignment was made by minimizing the RMS deviations of alpha carbons in the four P1 helices (positions p38 through p48) against respective positions in the Kv1.2 channel.
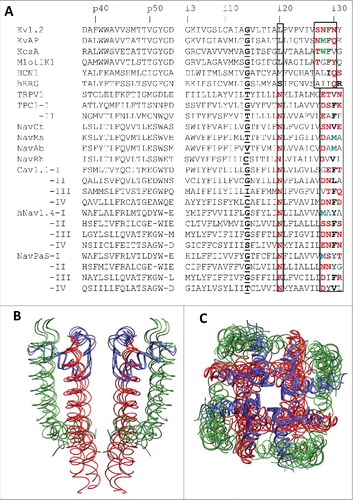
Figure 2. 3D Alignment and alpha carbon deviations in the inner helices. A, Inner helices in the closed channels MlotiK1, NavAb and KcsA. The channels are 3D aligned against the reference structure of the open Kv1.2 channel. Residues in positions i21, i20 and i29 (sticks) have the same orientations. Deviations of alpha carbons in the closed-state structures from the open-state Kv1.2 structure are small in positions i1 – i14 and steadily increase in positions i15 – i35, which are C-terminal to the gating-hinge residue. B, Inner helices in the open channels NavAb, NavMs and NavCt. The channels are 3D aligned against the reference structure of the open Kv1.2 channel. Residues in positions i21, i20 and i29 (sticks) have the same orientations. Deviations of the inner helix alpha carbons in the sodium channels from the open-state Kv1.2 are small in the N-halves of the helices. In the C-halves, the deviations increase (especially for the NavCt channel) due to variable aperture of the inner pore at the activation gate region. C, Deviations of the inner helix alpha carbons from the reference Kv1.2 structure for the NavRh channel calculated using the conventional (normal) sequence alignment () or an artificial alignment where the entire NavRh sequence is shifted one position right vs. Kv1.2. The shift results in large oscillations in the deviation plot. D, Intersubunit contacts between residues in positions Xi20 and (X-1)i29 in Kv1.2 and NavCt. Despite different nature and rather different mutual dispositions of the residues in positions i20 and i29, they have similar orientations and form open-state contacts Xi20: (X-1)i29.
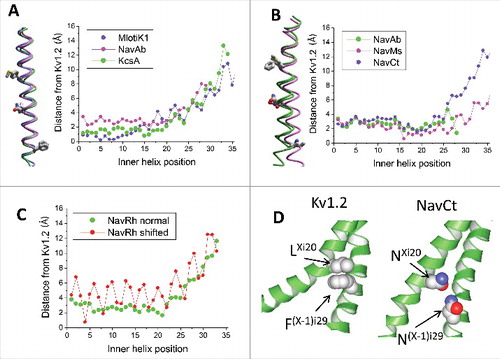
We further explored how sequence misalignment of S6s would affect deviations of individual alpha carbons from the reference structure. We shifted the sequence of the NavRh channelCitation7 one position right in the alignment and recalculated distances between the NavRh and Kv1.2 alpha carbons in the matching position of the shifted alignment. (The shift of the S6 sequence alignment does not affect the 3D alignment obtained by superposing the P1 helices). The misalignment caused large oscillations in the deviation plot (), which are in the sharp contrast with the smooth plot of deviations between matching atoms calculated for the alignment shown in . Thus, the smooth character of the deviation plots can serve as a criterion of the match between the sequence and 3D alignment. The fact that deviation plots in are smooth suggests the match of the sequence and 3D alignments between potassium and sodium channels.
Residues in position i20 and tetrads of polar-polar-hydrophobic-polar residues (p2hp motif) of potassium, sodium and calcium channels are involved in the open-state inter-subunit or inter-repeat contacts
To facilitate comparison of different channels, we use here a labeling scheme that is universal for P-loop channels (see Methods). The Kv1.2 channel has motif “SNFN” at the C-end of the inner helix and many other channels have the motif of p2hp residues (). Intriguingly, in the sequence alignment that corresponds to the 3D aligned structures, the p2hp motif in sodium and calcium channels is shifted by one position right vs. analogous motif in potassium channels (). The shift of the p2hp motif in the sequence alignment correlates with appearance of the conserved asparagines Ni20s in calcium and sodium channels (). Furthermore, there is a significant structural difference between the p2hp motifs in potassium and sodium channels.
In the open-gate Kv1.2 structure, hydrophobic phenylalanines FXi29 (the third position in the p2hp motif) form tight inter-subunit hydrophobic contact with hydrophobic leucines L(X-1)i20 (); both residues are conserved in many (but not all) potassium channels. The distance between beta-carbons of these residues is as small as 4.6 Å. In many other open-gate structures, residues Xi20 and (X-1)i29 also are in the close proximity. For example, in the cyclic nucleotide-gated channel HCN119 the methyl group of TXi20 forms a tight hydrophobic contact with I(X-1)i29. In the KvAP structure, which has a larger aperture of the open gate, side chains FXi29 diverge more from the pore axis and approach positions i13 and i17. However, the distance between beta-carbons in positions Xi20 and (X-1)i29 is also rather small (6.1 Å). It should be noted that the wide opening in the X-ray structure of the KvAP channelCitation11 may be due to the fact that the S4-S5 linker helices are disposed relative to the S6 helices, not like in the Kv1.21 or MlotiK120 channels, where the four S4-S5 linker helices form a cuff that limits divergence of the pore-lining S6 helices from the pore axis. The hERG channel has serine in position i20 where Kv channels have leucine. Furthermore, the hERG channel has motif AIIQ at the C-end of S6 where other P-loop channels have motif p2hp. In the available hERG structureCitation21 conformation of S6 segment (except the C-end) is close to that in Kv1.2 and residues SXi20 and I(X-1)i29 are in close proximity. However, these residues do not form strong stabilizing contact and the pore domain folding apparently is supported by other intersegment interactions. Probably, the structural differences between the Kv1.2 and hERG channels are partially due to the different specific contacts between residues in positions Xi20 and (X-1)i29.
In sodium and calcium channels positions i20 in all four repeats (subunits) are occupied by the exceptionally conserved asparagine residues, which are followed by a hydrophobic residue (usually leucine) in position i21. In the open-gate NavCt structure,Citation18 asparagines NXi20 and N(X-1)i29 approach each other to the distance, which allows inter-subunit H-bonding (). The distance between Cβ atoms in this pair is 8.3 Å. Inter-repeat contacts between highly conserved asparagines NXi20 and polar sidechains in positions (X-1)i29 are proposed to serve as important stabilizers of the open-state conformations of sodium and calcium channels.Citation22
Prokaryotic sodium channels NavAb and NavMs lack polar residues in positions i29. In the X-ray structure of the open-state NavMs,Citation4 asparagines NXi20 apparently lack polar contacts and form tight inter-segment contacts with sidechains of hydrophobic residues I(X-1)i26, M(X-1)i30 and VXo9. This fact seemingly argues against the proposition that inter-subunit open-state H-bonds involving Ni20 are common for sodium and calcium channels.Citation22 A closer inspection of the X-ray structure shows that the electronegative sidechain oxygen in NXi20 approaches the electronegative sulfur atom in M(X-1)i30. Such contact is energetically non-preferable and would bring destabilizing contribution to the open state energy. However, upon the 180° twist of the Ni20 side-chain torsion χ2, bonds Cγ-NH2 and Cγ = O in the amide group would swap. The two orientations of the amide group would be hardly distinguishable in the 2.45 Å resolution crystal structure,Citation4 but in the swapped conformations the H-bonds NXi20_NH—S_M(X-1)i30 would be formed. The NH—S hydrogen bonds are seen in X-ray structures of both small molecules and proteins.Citation23–26 Besides the likely H-bond NXi20—M(X-1)i30, the large sidechains M(X-1)i30 form close inter-subunit contacts with VXi27, IXi23, and GXi24. Furthermore, alanine residues A(X-1)i29, which are far from the conserved asparagines NXi20, form tight intersegment contacts with GXk10, MXk11, and AXk14. Thus, the lack of open-state H-bonding partners for asparagines NXi20 in position (X-1)29 is compensated by methionine residues M(X-1)i30 that likely form H-bonds with NXi20s.
In the open-state structure of NavAb,Citation6 asparagines NXi20 donate H-bonds to the sidechains of aspartates D(X-1)i28, which are the last C-terminal S6 residues resolved in the X-ray structure. Importantly, the C-ends of the inner helices adopt the 310 helix conformations, which are not seen in the X-ray structures of NavMs or closed-state NavAb. Obviously, in the open-state NavAb structure that lacks methionines M(X-1)i30, which are present in the closed-state NavAb, the C-ends of S6s adopt conformations that enable the conserved asparagines NXi20 to find the open-state H-bonding partner. The above analysis supports the proposition that highly conserved asparagines form inter-segment H-bonds that stabilize the open state of sodium channels.Citation22
Thus, the open-state structures in potassium channels are stabilized by intersubunit contacts LXi20: F(X-1)i29, whereas in sodium and calcium channels they are usually stabilized by H-bonds between the conserved asparagines NXi20 and a polar residue in positions (X-1)i29. Importantly, phenylalanine Fi29 in potassium channels and polar residues in the same position of sodium and calcium channels belong to the same p2hp motif, which is shifted in the aligned sequences of these channels (). It is hardly possible that so dramatic changes in the nature of the open-state stabilizing contacts between potassium channels on one hand and sodium/calcium channels on the other hand appeared in evolution as correlated mutations of individual amino acids. We suggest that the one-position shift of the p2hp motif in the sequence alignment is due to an insertion in the S6 sequences of sodium and calcium channels. To test this idea we considered other members of the P-loop channels family.
Residues Ni20s in TRPV1 and TPC1 channels
Asparagines NXi20, which are exceptionally conserved in sodium and calcium channels,Citation22 are seen in the sequences of TRPV and two-pore channels (). Furthermore, in these channels positions i21 and i30 contain hydrophobic residues, which are also present in sodium and calcium channels. Intriguingly, in the open-gate structure of TPC1, residues LXi21 and F(X-1)i30 form tight intersegment contacts with the distance of 5.1 Å between beta-carbons. These contacts are very similar to contacts between residues LXi20 and F(X-1)i29 in the Kv1.2 structure (). In contrast, the distance between beta carbons of the conserved asparagine NXi20 and a polar residue in position (X-1)i29 is as big as ∼10 Å. In the open-gate structure of the TRPV1 channel, the polar residues NXi20 and T(X-1)i29 also do not form contacts because the distance between respective beta carbons is as big as 11.7 Å, whereas hydrophobic residues MXi21 and V(X-1)i30 do form contacts with the distance of 8.3 Å between respective beta-carbons. Thus, despite the fact that TPC1 and TRPV1 channels contain asparagines Ni20, their open-state stabling contacts are formed between hydrophobic residues and in this respect these channels are more similar to Kv1.2 and other potassium channels. In other words, in the Kv1.2 and TPC1/TRPV1 channels, analogous contacts are formed by same-type residues, which are located in mismatched positions of the sequence alignment. In the 3D aligned structures, Kv1.2 residues Li20 and Fi29 have the same orientation as TPV1 residues Li21 and Fi30 (). In contrast, in the 3D aligned channels NavCt and TRPV1, asparagines Ni20 have different orientations, and polar residues in position i29 also have different orientations ().
Figure 3. Inner helices in the open-state Kv1.2, TPC1 and TRPV1 channels. A, Tight intersubunit contacts are formed by pairs of hydrophobic residues LXi20: F(X-1)i29 in Kv1.2 and LXi21: F(X-1)i30 in TPC1. In the sequence alignment these pairs of residues are shifted by one position (). B, Inner helices in the 3D-aligned structures of Kv1.2 and TPC1. The residues participating in the open-state contact (Li20 and Fi29 in Kv1.2 and Li21 and Fi30 in TPC) have similar orientations despite they are in mismatching positions of the sequence alignment (). C, Inner helices in the 3D-aligned structures of NavCt and TRPV1. Residues in positions i20 and i29 have different orientations despite they are in matching positions of the sequence alignment (). D, The π-Helix bulge in the TRPV1 inner helix. A residue in position i16 is red. E, Deviations of the inner helix alpha carbons from the Kv1.2 structure in the TPC1 and TRPV1 channels. When the straightforward sequence alignment was used (), large oscillations in the deviation plot appeared C-terminal to position i16 (open circles). Correction of the alignment by shifting the TPC1 and TRPV1 sequences one position right vs. the Kv1.2 sequence eliminated the oscillations (filled circles). F, In the TRPV1 channel, the sidechains of asparagines Ni20 donate H-bonds to the backbone carbonyls i16; these H-bonds stabilize the π-helix bulges. G, Distances between the inner-helix backbone atoms Hi and Oi-4, which are expected to form H-bonds in regular alpha helices. The alpha-helical H-bonds are formed in all residues in NavMs, but in the TRPV1 channel the pattern of H-bonds is broken in the middle of the inner helix where the π-helix bulge appears.
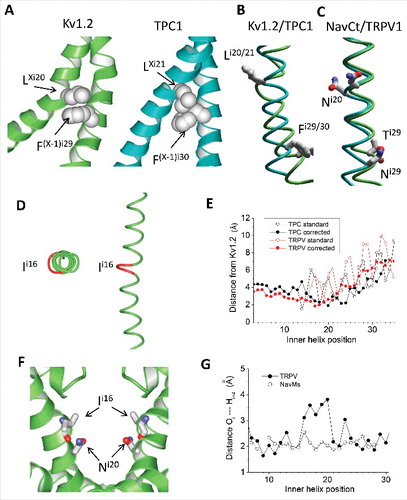
The shift of the open-state stabilizing contacts in TRPV1 and TPC1 channels correlates with the π-helix bulge N-terminal to Ni20
How the same-type residues within helical segments, which occur in mismatching positions of the sequence alignment, could form identical 3D contacts that apparently stabilize the open state? One possibility is that budges (π-helical elements) or constrictions (310 helical elements) may have appeared in the evolution. With this idea in mind we scrutinized the 3D structures of S6 helices in different channels. Indeed, unlike the MlotiK1, Kv1.2, NavAb, NavMs and NavCt channels, the TRPV1 and TPC1 channels contain π-helix budges () where the alpha-helical H-bonds are lacking () and π-helical H-bonds with the distances of ∼ 2.4 Å are formed. We calculated deviations of S6 alpha carbons in TRPV1 and TPC1 channels from matching atoms in the Kv1.2 channel using the sequence alignment shown in . We found that the deviation plots are smooth before position i16, but have large oscillation C-terminal to this position (open circles in ), indicating problems in this alignment. When we excluded position i16 from the TRPV1 and TPC1 sequences, the deviation lines become smooth (filled circles in ). In the TPC1 and TRPV1 structures, asparagines NXi20 do not approach residues in positions (X-1)i29. Unlike sodium and calcium channels, where side chains of asparagines Ni20 face away the pore axis, in the TRPV1 and TPC1 channels side-chains of asparagines NXi20 line the pore, extend in the extracellular direction, approach the “bachelor” backbone carbonyls O = C in positions i16 and can form H-bonds with these carbonyls (). Such specific side-chain to backbone interactions are known to stabilize the bulge structure.Citation27 It should be also noted that the polar side chains of asparagines Ni20 in the TPC1 and TRPV1 channels approach level i15 where sodium and calcium channels have pore-facing polar residues.
Hypothesis: Asparagines Ni20 appeared in evolution as an insertion
Comparison of available sequences and 3D structures allowed us to suggest that asparagines Ni20 appeared in evolution as insertions. In we propose a sequence alignment where many residues at the C-terminal thirds of the inner helices, which occur in matching positions, have similar physico-chemical properties. Besides well-aligned p2hp motifs in positions i28 – i31, the revised alignment shows large hydrophobic residues in positions i21 and i23, small residues (alanine or glycine) in position i24, and hydrophobic and usually large residues in position i26.
Figure 4. Alternative sequence alignments of the inner helices in P-loop channels. A, Corrected alignment in which asparagines in position i20 of sodium, calcium, TRPV and TPC channels are insertions vs. potassium channels. In this alignment, the boxed p2hp motifs (polar-polar-hydrophobic-polar residues) at the C-ends occur in matching positions. B, “Structure-based” sequence alignment corresponding to the 3D aligned structures in which all residues are in 3D-matching positions and form similar inter-subunit contacts that stabilize the open state. The insertions are placed in position i16, which forms a bulge in the TPC1 structure.
In the TRPV1 and TPC1 channels, the insertion is accommodated by formation of the π-helical bulge, which is stabilized by the sidechain-backbone H-bond involving asparagine Ni20. In the X-ray structures of prokaryotic sodium channels, asparagines Ni20 face away the pore, the S6 helices have alpha-helical conformations and lack bulges, while their cytoplasmic thirds C-terminal to position i19 are reoriented by a ∼ 100° twist. This dramatic reorientation should be accompanied by switching of many contacts between the C-terminal thirds of S6s with neighboring S5, L45 and S6 helices.
The revised sequence alignment with insertions/deletions at position i20 () may reflect the evolutionary history of P-loop channels. However, neither this alignment nor the conventional alignment () is completely suitable for homology modeling. Indeed, the sequence alignment between template and query proteins in homology modeling usually implies that residues in the matching positions of the sequence alignment should also have matching (similar) orientations of side chains in the 3D-aligned structures and would form similar intersegment contacts. However, despite the fact that in both alignments ( and ) the NavCt and TPC1/TRPV1 channels have polar residues in positions i20 and i29, orientations of these residues are different (). This difference reflects different ways by which the Ni20 insertion is accommodated. The structure-based alignment of bacterial sodium channels and TPC1 is shown in . In this alignment all residues in matching positions have the same 3D orientations. The insertion is proposed at positioned i16, where the TPC1 structure has the π-helix bulge.
Channels with and without π-helical bulges should be considered as two distinct template categories and prior to homology modeling a decision should be made to which category the query protein may belong. Presently, it remains unclear why in the sodium and calcium, but not in TPC1 and TRPV1 channels the bulges disappeared and C-terminal thirds of S6s are twisted. All these channels permeate hydrated cations and all they have a ring of the pore-facing hydrophilic residues below the selectivity filter. In calcium and sodium channels these are asparagine and/or serine residues in positions i15, whereas in the TPC1 and TRPV1 channels these are asparagine residues in positions i20 whose sidechains extend towards the selectivity filter and occur at approximately the same level of the pore as hydrophilic residues in positions i15 of calcium and sodium channels (). Notably, TPC1 and TRPV1 channels have in positions i15 hydrophobic or predominately hydrophobic (tyrosine) residues. Better understanding of structural causes by which the asparagine insertions cause π-helix bulges or twists of the inner helix C-terminal thirds is necessary to make such decisions. A comprehensive analysis of multiple inter-segment contacts of the inner helices in different P-loop channels requires a stand-alone study.
Modeling artificial analogues of the TRPV1 channel without π-helical bulges
Possible structural consequences of the proposed insertions/deletions in the sequence alignment () can be explored in models of artificial channels, which we did not find in databases. First, we modeled an analogue of the TRPV1 channel (TRPV1*) in which asparagines Ni20 were deleted. Intensive Monte Carlo energy minimizations yielded the structure, which is very similar to the original TRPV1 X-ray structure (RMSD of alpha carbons is 0.77 Å) but the inner helices adopted classical alpha-helical conformations. In other words, removal of the asparagines caused relaxation of the π-helical bulges without reorientation of other residues (). The major difference was found for threonines Ti16, which in the X-ray structure are involved in the S6 budges. In the TRPV1* channel, threonines Ti16 partially lost their contacts with the outer-helix residues in positions Xo6 and Xo9 of the same subunit, as well as with inner-helix residues in positions (X-1)i19 and (X-1)i26 of the adjacent subunit. We further have built a TRPV1 model without the S6 bulges (TRPV1**) by imposing alpha-helical constraints between backbone atoms and facilitating reorientation of the C-terminal third of S6 by imposing H-bonds between side chains of NXi20 and N(X-1)i29. The Monte Carlo-minimized structure of TRPV1** approached the NavCt conformation () and the RMS deviation of S6 alpha carbons from the Kv1.2 reference structure decreased from 3.6 to 2.8 Å. In this structure, threonines Ti16 also lost their intersegment contacts. Certainly, other contacts contribute to the open- and closed-state energies of P-loop channels.
Figure 5. Models of artificial analogs of the TRPV1 channel. A, Comparison of the open-state structures of HCN1, TRPV1 and TRPV*, the artificial analogue of TRPV1 with the Ni20 deletion. Due to the helix bulge in TRPV1, residues MXi21 and V(X-1)i30 form a contact, which is similar to the contact between MXi20 with I(X-1)i29 in the HCN1 channel. Deletion of asparagine Ni20 in the TRPV* model caused disappearance of the bulge and appearance of contact MXi20: V(X-1)i29, which matches contact MXi20: V(X-1)i29 in HCN1. B, Comparison of the open-state structures of NavCt (left), TRPV1 (middle) and TRPV1**, the artificial analogue of TRPV1 with the twisted C-terminal thirds of the inner helices (right). In the NavCt and TRPV** channels, residues in positions Xi20 and (X-1)i29 form intersubunit open-state contacts. In the TRPV1 channel the inner helix has a π-helix bulge and respective contact is lacking. Imposing the alpha-helical conformations of the inner helices in the TRPV1** channel resulted in formation of the H-bond between NXi20 and T(X-1)i29.
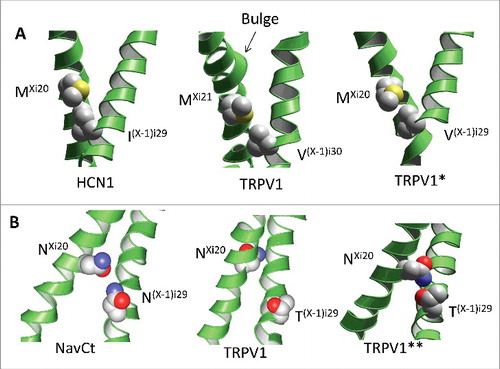
Although these models are speculative, they may have analogues among P-loop channels. For example, hyperpolarization-activated cyclic nucleotide-gated channel HCN1 has methionine rather than asparagine in position i20 (). The S6 helix of HCN1 has no bulge and, as a result, orientation of Mi20 matches that of Mi21 in TRPV1.Citation19 In other words, in HCN1, structure of the S6 helix and contacts Xi20 – (X-1)i29 correspond to our TRPV* model ().
Comparison of Cav1.1, NavPaS and Nav1.4
Atomic-scale structures of many pharmacologically important sodium and calcium channels in different functional states are still lacking. Therefore, structural interpretations of various experimental data including, but not limited to the channel gating, drug action and channelopathy mutations are analyzed basing on available structures of relative channels. Our results show that the latter structures may significantly differ in channels with homologues sequences. Recently, structures of the rabbit Cav1.1,Citation9 electric eel Nav1.428 and cockroach NavPaSCitation8 channels have been published (). These channels contain asparagines Ni20 (except K3i20 in NavPaS) and usually have the p2hp motifs at the C-ends of S6s.
Figure 6. Different structures of the inner helices in Cav1.1, NavPaS and Nav1.4. A, Cytoplasmic view of the inner helices in the 3D-aligned Nav1.4 (blue), NavPaS (green) and Cav1.1 (red) channels. Space-filled are the Cα-Cβ bonds of asparagines Ni20. These bonds face the pore in NavPaS, but face away the pore axis in Cav1.1 and Nav1.4 (except for N3i20 in IIIS6). B, Side view at the superimposed helices IS6 of NavPaS, Nav1.4 and Cav1.1. The side chains in positions i12, i20 and i29 are shown. The side chains in position i12 have similar orientations, while orientations of residues in positions i20 and i29 are different. C-E, Distances between backbone atoms Oi and Hi+4 in the inner helices of Cav1.2 (C), NavPaS (D) and Nav1.4 (E). Data for repeats I, II, III, and IV are shown black, red, green and blue, respectively. In Cav1.1 (especially in repeat IV), most of the distances correspond to the classical alpha-helix. In NavPaS, but not in Cav1.1, the pattern of alpha-helical H-bonds is broken in the middle of S6. π-Helical H-bonds are formed in positions i15 – i20 of NavPaS, but not in Cav1.1. Characteristics of helical H-bonds of repeats I, II and IV in Nav1.4 are similar to those in Cav1.1, whereas in repeat III they are similar to those NavPaS (E).
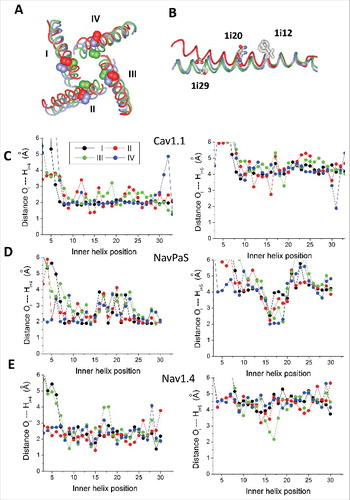
The Cav1.1 structure is largely asymmetric and the inner helices in repeats II and IV diverge from the pore axis, resembling available open-gate structures. In repeats I and III the inner helices are in the closed-gate conformation. Correspondingly, in interfaces II/I and IV/III the distances between beta-carbons in positions Xi20 and (X-1)i29 are below 8 Å, implying that the pairs of polar residues, which do not face the pore, can form inter-repeat H-bonds. In interfaces III/II and IV/I the distances between beta carbons in respective positions are above 11 Å. Although the inner helices are not perfect, there is no sign of π-helical bulges ().
In the closed-state NavPaS structure, H-bonds between residues in positions Xi20 and (X-1)i29 are not possible. The inner helices have π-helical bulges, disrupted alpha-helical H-bonds in positions i16 – i22 and π-helical H-bonds in positions i15 – i20 (), whereas asparagines in position i20 face the pore (). By these features the NavPaS inner helices are remarkably similar to those in the TRPV1 and TPC1 channels. The plot of alpha carbons deviations from the Kv1.2 reference structure, which is calculated using the conventional sequence alignment, has oscillations C-terminal to position i15 (data not shown). The oscillations can be eliminated by excluding position i16 from the alignment. In contrast to NavPaS, the conventional alignment of Cav1.1 with Kv1.2 yielded smooth lines of the alpha carbon deviations, but oscillations appeared when residues in position i16 were excluded from the Cav1.1 sequence. Thus, the eukaryotic NavPaS and Cav1.1 channels have different organization of the inner helices, different patterns of the pore-facing residues, and different intersegment contacts. The NavPaS channel is non-functionalCitation29 and we cannot rule out that this is due to the described peculiarities of the inner helices structure.
The Nav1.4 structure is also asymmetric, the distances between beta-carbons in positions Xi20 and (X-1)i29 are 7.4, 6.2, 8.3 and 11.4 Å. The asymmetry is partially due to different structural organizations of the inner helices, in repeats I, II and IV where the segments are entirely alpha-helical as in Cav1.1 and prokaryotic sodium channels (). But repeat III has π-helical H-bonds between positions i17 and i12 and corresponding break of the alpha-helical H-bonding pattern. Plots of alpha-carbon deviations also revealed this structural difference: oscillations in the C-half are seen only in repeat III if the template structure is Kv1.2 or Cav1.1. If the NavPaS structure is taken as the template, the oscillations appear in repeats I, II and IV (data not shown).
As a result of the structural differences, the i20 orientations in repeats I, II and IV are the same in Nav1.4 and Cav1.1, whereas in repeat III the i20 orientation in Nav1.4 is intermediate between that seen in the NavPaS and Cav1.1 channels ( and ). Asparagine N3i29 in repeat III does not approach N2i20, but forms an H-bond with a serine residue in the linker helix of the same repeat. Thus, recent structures of eukaryotic channels show alpha-helical organization of S6s in Cav1.1, π-helical bulges in the middle of all four S6s in NavPaS, and a bulge in IIIS6 of Nav1.4.
Conclusions
The sequence and 3D alignments of P-loop channels suggest that conserved asparagines Ni20 in the inner helices of sodium, calcium, TRPV and TPC channels appeared in evolution as insertions. In the TRPV1, TPC1 and NavPaS channels, the insertions are accommodated as π-helix bulges that preserve contacts of residues beyond the budges. In other channels of known 3D structures the insertions apparently caused reorientation of the S6 C-terminal thirds. These differences should be considered in homology modeling of P-loop channels and structural interpretation of experimental data.
The causes of different structural accommodations of the Ni20 insertion in different channels remain unclear. They may depend on sequence-related properties of individual channels, the channels functional states, or specific environment. Therefore, the structural interpretation of experimental data obtained on the channels of unknown structure may or may not be valid when structures of apparently homologous channels are used as templates. Comprehensive analyses of various experimental data may help resolve this problem.
Methods
To compare 3D orientations of residues in P-loops and inner helices, we used the alignment elaborated and used in our previous studies (). Above the alignment, residue labels that are universal for P-loop channels are shown.Citation3 A label designates repeat number, segment type (k, linker-helix S4-S5; o, Outer helix (S5); p, P-loop; i, Inner helix (S6) and relative residue number in the segment. For example, F4i15 designates phenylalanine in position 15 of the inner helix in the fourth repeat. At the extracellular view of eukaryotic sodium and calcium channels repeats I, II, III and IV are arranged clockwise.Citation30 To describe some inter-repeat contacts in these channels, as well as inter-subunit contacts in homotetrameric P-loop channels, we assign prefix “X” for a residue label in the given repeat and prefixes (X-1) and (X+1), respectively, for its clockwise and anticlockwise subunit/repeat neighbors at the extracellular view.
Comparison of X-ray and cryo-EM structures of different P-loop channels in the open and closed states shows that four P1 helices are the most structurally conserved segments whose mutual dispositions only minimally varies between channels. Therefore, to superimpose 3D structures of different channels (obtain their 3D alignment), we used the X-ray structure of the potassium channel Kv1.2 (1) as a reference, kept the channel structures rigid, and minimized the RMS deviation between alpha carbons in Kv1.2 and another channel in positions p38 through p48 of the four P1 helices.
Modeling of artificial analogues of TRPV1 was performed using the ZMM program package.Citation31
Author contributions
D.B.T. Proposed the research, compared structures and performed calculations. D.B.T. and B.S.Z. analyzed results and wrote the manuscript.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
The study was supported by the RAS program “Molecular and Cell Biology” (a grant to D.B.T.) and by the Russian Foundation for Basic Research (grant #17-04-00549 to B.S.Z).
References
- Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science (New York, NY. 2005;309:897–903. doi:10.1126/science.1116269.
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science (New York, NY. 1998;280:69–77. doi:10.1126/science.280.5360.69.
- Zhorov BS, Tikhonov DB. Potassium, sodium, calcium and glutamate-gated channels: pore architecture and ligand action. J Neurochem. 2004;88:782–99. doi:10.1111/j.1471-4159.2004.02261.x. PMID:14756799.
- Sula A, Booker J, Ng LC, Naylor CE, DeCaen PG, Wallace BA. The complete structure of an activated open sodium channel. Nat Commun. 2017;8:14205. doi:10.1038/ncomms14205. PMID:28205548.
- Korkosh VS, Zhorov BS, Tikhonov DB. [Molecular Evolution of Ion Channels: Amino Acid Sequences and 3d Structures]. Zhurnal Evoliutsionnoi Biokhimii i Fiziologii. 2016;52:26–33. PMID:27220237.
- Lenaeus MJ, Gamal El-Din TM, Ing C, Ramanadane K, Pomes R, Zheng N, Catterall WA. Structures of closed and open states of a voltage-gated sodium channel. Proc Natl Acad Sci U S A. 2017;114:E3051–E60. doi:10.1073/pnas.1700761114. PMID:28348242.
- Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L, Wang J, Hasegawa K, Kumasaka T, He J, et al. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature. 2012;486:130–4. PMID:22678295.
- Shen H, Zhou Q, Pan X, Li Z, Wu J, Yan N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science (New York, NY. 2017;355(6328), eaal4326. doi:10.1126/science.aal4326.
- Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q, Yan N. Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature. 2016;537:191–6. doi:10.1038/nature19321. PMID:27580036.
- Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–8. doi:10.1038/nature10238. PMID:21743477.
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi:10.1038/nature01580. PMID:12721618.
- Huber I, Wappl E, Herzog A, Mitterdorfer J, Glossmann H, Langer T, Striessnig J. Conserved Ca2+-antagonist-binding properties and putative folding structure of a recombinant high-affinity dihydropyridine-binding domain. Biochem J. 2000;347 Pt 3:829–36. doi:10.1042/bj3470829. PMID:10769189.
- Lipkind GM, Fozzard HA. KcsA crystal structure as framework for a molecular model of the Na(+) channel pore. Biochemistry. 2000;39:8161–70. doi:10.1021/bi000486w. PMID:10889022.
- Zhorov BS, Folkman EV, Ananthanarayanan VS. Homology model of dihydropyridine receptor: implications for L-type Ca(2+) channel modulation by agonists and antagonists. Archives Biochem Biophys. 2001;393:22–41. doi:10.1006/abbi.2001.2484.
- Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–12. doi:10.1038/nature12822. PMID:24305160.
- Guo J, Zeng W, Chen Q, Lee C, Chen L, Yang Y, Cang C, Ren D, Jiang Y. Structure of the voltage-gated two-pore channel TPC1 from Arabidopsis thaliana. Nature. 2016;531:196–201. doi:10.1038/nature16446. PMID:26689363.
- Korkosh VS, Zhorov BS, Tikhonov DB. Analysis of inter-residue contacts reveals folding stabilizers in P-loops of potassium, sodium, and TRPV channels. European Biophys J. 2016;45:321–9. doi:10.1007/s00249-015-1098-6.
- Tsai CJ, Tani K, Irie K, Hiroaki Y, Shimomura T, McMillan DG, Cook GM, Schertler GF, Fujiyoshi Y, Li XD. Two alternative conformations of a voltage-gated sodium channel. J Mol Biol. 2013;425:4074–88. doi:10.1016/j.jmb.2013.06.036. PMID:23831224.
- Lee CH, MacKinnon R. Structures of the Human HCN1 hyperpolarization-activated channel. Cell. 2017;168:111–20 e11. doi:10.1016/j.cell.2016.12.023.
- Clayton GM, Altieri S, Heginbotham L, Unger VM, Morais-Cabral JH. Structure of the transmembrane regions of a bacterial cyclic nucleotide-regulated channel. Proc Natl Acad Sci U S A. 2008;105:1511–5. doi:10.1073/pnas.0711533105. PMID:18216238.
- Wang W, MacKinnon R. Cryo-EM structure of the open human ether-a-go-go-related K+ Channel hERG. Cell. 2017;169:422–30 e10. doi:10.1016/j.cell.2017.03.048.
- Tikhonov DB, Bruhova I, Garden DP, Zhorov BS. State-dependent inter-repeat contacts of exceptionally conserved asparagines in the inner helices of sodium and calcium channels. Pflugers Archiv. 2015;467:253–66. doi:10.1007/s00424-014-1508-0. PMID:24728659.
- Allen FH, Bird CM, Rowland RS, Raithby PR. Hydrogen-bond acceptor and donor properties of divalent sulfur (Y-S-Z and R-S-H). Acta Cryst. 1997;B53:696–701. doi:10.1107/S0108768197002644.
- Biswal HS, Wategaonkar S. Nature of the N-H…S hydrogen bond. J Phys Chem. 2009;113:12763–73. doi:10.1021/jp907658w.
- Chung WP, Dewan JC, Walters MA. Models of lysine-cysteine hydrogen bonding in metallothionein: hydrogen bonding between ammonium and benzenethiolate in [(C6H11)2 NH2]2[Co(SC6H5)4]. J Am Chem Soc. 1991;113:525–30. doi:10.1021/ja00002a021.
- Ma Q, Zhao X, Nasser Eddine A, Geerlof A, Li X, Cronan JE, Kaufmann SH, Wilmanns M. The Mycobacterium tuberculosis LipB enzyme functions as a cysteine/lysine dyad acyltransferase. Proc Natl Acad Sci U S A. 2006;103:8662–7. doi:10.1073/pnas.0510436103. PMID:16735476.
- Kumar P, Bansal M. Dissecting pi-helices: sequence, structure and function. FEBS J. 2015;282:4415–32. doi:10.1111/febs.13507. PMID:26370783.
- Yan Z, Zhou Q, Wang L, Wu J, Zhao Y, Huang G, Peng W, Shen H, Lei J, Yan N. Structure of the Nav1.4-beta1 Complex from Electric Eel. Cell. 2017;170:470-82 e11. doi:10.1016/j.cell.2017.06.039.
- Moignot B, Lemaire C, Quinchard S, Lapied B, Legros C. The discovery of a novel sodium channel in the cockroach Periplaneta americana: evidence for an early duplication of the para-like gene. Insect Biochem Mol Biol. 2009;39:814–23. doi:10.1016/j.ibmb.2009.09.006. PMID:19800971.
- Dudley SC Jr., Chang N, Hall J, Lipkind G, Fozzard HA, French RJ. mu-conotoxin GIIIA interactions with the voltage-gated Na(+) channel predict a clockwise arrangement of the domains. J General Physiol. 2000;116:679–90. doi:10.1085/jgp.116.5.679.
- Garden DP, Zhorov BS. Docking flexible ligands in proteins with a solvent exposure- and distance-dependent dielectric function. J computer-Aided Mol Design. 2010;24:91–105. doi:10.1007/s10822-009-9317-9.
- Tikhonov DB, Zhorov BS. Architecture and pore block of eukaryotic voltage-gated sodium channels in view of NavAb bacterial sodium channel structure. Mol Pharmacol. 2012;82:97–104. doi:10.1124/mol.112.078212. PMID:22505150.