
ABSTRACT
A fundamental mechanism that drives the propagation of electrical signals in the nervous system is the activation of voltage-gated sodium channels. The sodium channel subtype Nav1.7 is critical for the transmission of pain-related signaling, with gain-of-function mutations in Nav1.7 resulting in various painful pathologies. Loss-of-function mutations cause complete insensitivity to pain and anosmia in humans that otherwise have normal nervous system function, rendering Nav1.7 an attractive target for the treatment of pain. Despite this, no Nav1.7 selective therapeutic has been approved for use as an analgesic to date. Here we present a summary of research that has focused on engineering peptides found in spider venoms to produce Nav1.7 selective antagonists. We discuss the progress that has been made on various scaffolds from different venom families and highlight the challenges that remain in the effort to produce a Nav1.7 selective, venom-based analgesic.
Voltage-gated sodium channels (VGSC) are present in all excitable cells including cardiac and skeletal muscle cells and central and peripheral neurons. In neuronal cells, sodium channels are responsible for amplifying sub-threshold depolarizations and generating the rapid upstroke of the action potential. As such, sodium channels are essential to the initiation and propagation of electrical signals in the nervous system. Aberrant sodium channel function is thought to underlie a variety of medical disorders [Citation1] including epilepsy [Citation2], arrhythmia [Citation3] myotonia [Citation4], and pain [Citation5].
Sodium channels are typically a complex of various subunits, the principal one being the pore-forming alpha subunit, which alone is sufficient for function. Nine members of the family of voltage-gated sodium channel (VGSC) alpha subunits exist in humans, Nav1.1–Nav1.9. The Nav1.x subfamily can be pharmacologically subdivided into tetrodotoxin (TTX) sensitive or TTX resistant. Nav1.7 (also named as PN1, SCN9A, or hNE) is TTX sensitive and is primarily expressed in peripheral sympathetic and sensory neurons [Citation6]. Nav1.7 is robustly expressed along the peripherally and centrally directed C-fibers and at nodes of Ranvier in a subpopulation of Aδ-fibers. Peripheral terminals of DRG neurons within skin (intraepidermal nerve fibers) exhibit robust Nav1.7 immunolabeling, as do the central projections of DRG neurons in the superficial lamina of the spinal cord dorsal horn (Black et al., 2012). Given the widespread expression along peripheral nerve fibers, Nav1.7 is likely a major contributor to electrogenesis, axonal conduction, and presynaptic neurotransmitter release in peripheral nociceptors [Citation7].
Human genetic studies have strongly implicated Nav1.7 function in human pain sensation. In man, gain of function mutations of Nav1.7 have been linked to primary erythermalgia (PE), a disease characterized by burning pain and inflammation of the extremities [Citation8] and paroxysmal extreme pain disorder (PEPD) [Citation9]. Consistent with these observations, nonselective sodium channel blockers lidocaine, mexiletine, and carbamazepine can provide symptomatic relief in these painful disorders [Citation9,Citation10]. Single nucleotide polymorphisms in the coding region of SCN9A have been associated with increased nociceptor excitability and pain sensitivity [Citation11]. For example, a polymorphism rs6746030 resulting in R1150W substitution in human Nav1.7 has been associated with osteoarthritis pain, lumbar discectomy pain, phantom pain, and pancreatitis pain [Citation12]. Dorsal root ganglion (DRG) neurons expressing the R1150W Nav1.7 display increased firing frequency in response to depolarization [Citation13]. A disabling form of fibromyalgia has been associated with SCN9A sodium-channel polymorphism rs6754031, indicating that some patients with severe fibromyalgia may also have a DRG sodium channelopathy [Citation14].
Loss-of-function mutations of SNC9A in humans cause congenital indifference to pain, a rare autosomal recessive disorder characterized by a complete insensitivity to painful stimuli [Citation15–26]. Despite this, other sensory modalities in these patients are largely intact with some patients that show deficits in olfaction [Citation21,Citation25–28]. These patients generally have normal cognitive, motor, and autonomic function [Citation15–28] with the notable exceptions of a patient with hypohydrosis [Citation29], and 2 patients with hearing loss, bone dysplasia, and hypoguesia [Citation30].
Mice with global knockout of SCN9A are insensitive to peripheral injection of nonselective sodium channel activators or intra-dermal histamine injection and did not develop complete Freund’s adjuvant-induced thermal hyperalgesia [Citation31]. Targeted deletion of the SCN9A gene in nociceptive neurons causes a reduction in mechanical and thermal pain thresholds and reduction or abolition of inflammatory pain responses [Citation32]. Ablating Nav1.7 gene expression in all sensory neurons abolishes mechanical pain, inflammatory pain, and reflex withdrawal responses to heat. Deleting SCN9A in both sensory and sympathetic neurons abolishes mechanical, thermal, and neuropathic pain, and recapitulates the pain-free phenotype seen in humans with SCN9A loss-of-function mutations [Citation33].
The evidence presented above suggests that Nav1.7 inhibitors or blockers may be useful in the treatment of a wide range of pain associated with various disorders. Although some progress has been made in identifying small molecules with selectivity for Nav1.7 over other VGSC isoforms these efforts have yet to yield a viable therapeutic, and research has expanded focus to include large molecules [Citation34,Citation35]. A rich source of good starting points for Nav1.7 drug discovery are peptides found in the venom of spiders [Citation36].
Numerous publications have extensively reviewed the spider venom structure, function, and therapeutic potential [Citation34,Citation36–43]. In brief, spider venoms contain complex mixtures of compounds, with a high percentage of which are peptides of a broad range of functions [Citation41]. Many of these peptides are small, disulfide-rich toxins that impact voltage-gated sodium channel function [Citation37]. Because these molecules serve to incapacitate prey and dissuade predation, some of them have evolved to target a broad array of sodium channels that are essential for vital functions [Citation40,Citation42].
In this review, we focus on engineering strategies designed to maintain toxin Nav1.7 activity while reducing activity against Nav isoforms critical for fundamental physiological functions – the introduction of mutations that maintain or favor toxin interaction with Nav1.7 while diminishing toxin affinity for other Nav isoforms. Numerous academic and commercial laboratories have adopted this approach. Here we discuss the most salient advances in engineering key toxins from three NaSpTx families with well documented Nav1.7 activity, family I, exemplified by tarantula toxins such as Huwentoxin-IV (HwTx-IV) [Citation44] and Phrixotoxin 3 [Citation45], family II, containing Pn3a [Citation46], and family III, represented by Protoxin-II [Citation47] and Phrixotoxin-I [Citation45].
NaSpTx family I
Family 1 is a family of peptides isolated from the venom of tarantulas. They consist of 33–35 amino acid residues with three disulfide bridges that form an inhibitor cystine knot (ICK) motif. The best-characterized Nav1.7 blocker from this family is Huwentoxin-IV.
Huwentoxin-IV
Huwentoxin-IV (HwTX-IV, μ-TRTX-Hh2a) is a 35 amino acid peptide component of Chinese bird spider (Haplopelma Schmidti; also known as Ornithoctonus huwena, and Selenocosmia huwena [Citation44,Citation48]) venom. It is an inhibitory cysteine knot (ICK) peptide that blocks Nav1.7 and other TTX-sensitive voltage-gated sodium channels with an IC50 of ~30 nM [Citation44] and functions as a gating modifier by trapping the voltage sensor of domain II (VSD2) in an inward, closed conformation [Citation49]. Early mutagenesis studies on the Nav1.7 VSD identified key residues in the S1-S2 and S3-S4 regions of VSD2 that are critical for HwTX-IV’s Nav1.7 activity. Of these positions, mutation of E818 had the most dramatic impact on HwTX-IV’s ability to inhibit Nav1.7 activation. Subsequently, many studies focused on HwTX-IV have helped map regions of the toxin that mediate its interaction with Nav1.7.
Revell et al. [Citation50]. and Minassian et al. [Citation51] performed alanine scanning mutagenesis on non-cysteine positions in HwTX-IV. Both identified positions E1 and E4 as sites where the potency of HwTX-IV could be improved, and F6, R26, W30, K32, and Y33 as residues that are critical for toxin activity on Nav1.7. Minassian et al. [Citation51] also demonstrated that mutation of positions W30 and K32, which showed the most robust impairments of HwTX-IV activity in both studies, did not disrupt toxin structure. One discrepancy between these datasets is that Minassian et al. saw >10-fold decrease in Nav1.7 potency with alanine substitution at position I35, whereas Revell et al. saw no change. This may be related to the addition of an additional gly-lys to the c-terminus of Minassian et al.’s recombinantly produced peptide, whereas Revell et al. truncated their synthetic peptide at I35 in a carboxamide, suggesting a critical role for the C-terminus in HwTX-IV’s interface with Nav1.7. In addition, Minassian et al. identified positions P11, D14, L22, and S25 as being important for Nav1.7 activity – positions which were not interrogated by Revell et al. [Citation50,Citation51]. Minassian et al. also counter-screened the alanine scanning library of HwTX-IV variants against Nav1.2 and found a large degree of overlap between residues critical for Nav1.7 and Nav1.2 activity, with all positions flagged for Nav1.7 also being important for Nav1.2 activity except I35. Interestingly, mutation of positions R26 and K27 to alanine was shown to impact Nav1.2 potency to a greater degree than seen on Nav1.7, suggesting these residues may be involved in isoform-specific VGSC interactions.
Based upon the alanine scan data, Revell et al. implemented a strategy of substituting a small subset of amino acids with diverse physiochemical properties in positions E1, E4, F6, and Y33. This resulted in the synthesis of [E1G, E4G, Y33W]-HwTX-IV which showed >40-fold improvement in Nav1.7 potency while maintaining selectivity against Nav1.5 [Citation50]. Subsequent studies demonstrated that these mutations in a recombinantly produced, unamidated peptide did not perturb its structure. In addition, the broader selectivity of this peptide was investigated and while activity was maintained against most TTX-sensitive Nav isoforms, [E1G, E4G, Y33W]-HwTX-IV was less potent against Nav1.4 [Citation52].
A more extensive mutagenesis effort by Neff et al [Citation53] produced a large library of recombinant, unamidated peptides by methodically substituting amino acids at all non-cysteine positions with a focus on improving Nav1.7 potency and selectivity against Nav1.2. For Na1.7, this study reinforced the importance of the F6, R26, W30, K32, and Y33 positions in maintaining Nav1.7 activity. In addition, this study aligned well with the Minassian study in showing that P11, D14, S25, and I35 were important while also identifying I5, K27, and T28 as important determinants of Nav1.7 potency. The study also confirmed the importance of all residues except P11, L22, and S25 for inhibition of Nav1.2 as well as identifying and R26, K27, and I35 as fundamental to Nav1.2 activity [Citation53], Positions E1 and E4 were flagged by all three mutagenesis studies as positions where the Nav1.7 potency of HwTX-IV could be increased [Citation50,Citation51,Citation53]. These changes did not improve selectivity against Nav1.2, however, and in fact Neff et al. found that increased basicity at these positions preferentially increased Nav1.2 potency [Citation53].
Neff et al. also tried to combine mutations that improved potency against Nav1.7 and/or reduced activity against Nav1.2. [E1N, E4R, R26K, Q34S]-HwTX-IV was 10-fold more potent than recombinant huwentoxin against Nav1.7 but did not confer any significant selectivity when tested against Nav1.1 – Nav1.6. In contrast, [E1N, R26K, Q34S, G36I]-HwTX-IV only marginally improved Nav1.7 potency but decreased activity against Nav1.2 and Nav1.3 by 10-fold. Potency of the [E1N, E4R, R26K, Q34S]-HwTX-IV was improved an additional 6-fold by grafting R26T into this scaffold, while maintaining the selectivity window against Nav1.2 [Citation53].
Deng et al. [Citation54] produced targeted mutations in Huwentoxin by disrupting basic residues as well as adjacent positions in the C-terminus of the peptide. They found a minimal effect on activity against TTX-sensitive sodium currents in DRGs with K27A, and a reduction of activity by 12, 24, and 260-fold for T28D, R29A, and Q34D, respectively [Citation54]. Neff et al. also found that R29A and Q34D diminished Nav1.7 activity but did not test T28D. They did, however, test T28E, which showed a robust reduction in activity [Citation53]. While these data are not all directly comparable, they suggest a role for all these positions in maintaining HwTX-IV’s activity.
A notable feature of HwTX-IV (and other gating modifier toxins) is the cluster of hydrophobic residues or “hydrophobic patch” on one face of the molecule which is surrounded by a ring of charged amino acids. This face of the molecule is hypothesized to play a role in toxin membrane partitioning and interaction with charged phospholipid head groups in addition to making specific interactions with the channel [Citation55]. To test whether increased hydrophobicity improved HwTX-IV potency, Agwa et al. produced a modified version of the Revell peptide, [E1G, E4G, F6W, Y33W]-HwTX-IV, termed gHwTX-IV, with increased membrane binding affinity. This peptide was, however, approximately 15-fold less potent than [E1G, E4G, Y33W]-HwTX-IV [Citation55] suggesting that F6 mediates a critical, more specific contact beyond facilitating membrane partitioning.
Neff et al. showed that increased hydrophobicity at positions E4, A8, Y33, Q34, and G36 improved Nav1.7 potency [Citation53]. These residues flank positions that have been hypothesized to act as a membrane anchor [Citation53,Citation55,Citation56], thus strengthening the correlation between hydrophobicity and toxin potency. The mean impact of substituting hydrophobic residues into broad segments of HwTX-IV, however, was not significant suggesting that increased lipid affinity only consistently confers benefit in or around the hydrophobic patch [Citation53].
A subsequent effort by Agwa et al. [Citation57] altered positively charged residues (R26, K27, R29) in loop 4 of gHwTX-IV. While the lipid affinity of these peptides was reduced, the Nav1.7 potency of [R26A]-gHwTX-IV was increased ~4-fold. In addition, this peptide showed a markedly improved selectivity against Nav1.1 (9-fold), Nav1.2 (41-fold), and Nav1.6 (56-fold) while maintaining selectivity against Nav1.3, Nav1.4, and Nav1.5. Despite a lack of improvement in Nav1.7 potency [K27A] and [R29A]-gHwTX-IV also had better selectivity profiles, with [R29A]-gHwTX-IV showing a prominent increase in selectivity against Nav1.1 and Nav1.6 (>50-fold). In addition, Agwa et al. showed that intraplantar injection of [R26A]-gHwTX-IV reduced OD-1-induced nocifensive behaviors in mice [Citation57]. While these data demonstrate proof of principle in terms of target engagement and pharmacodynamic effect, additional studies will be needed to assess g-HwTX-IV variant tolerability and ability to access sensory fibers when administered systemically.
The data from these mutagenesis studies have led to the proposal of multiple models of how HwTX-IV interacts with Nav1.7. Minassian et al. proposed a model based upon their alanine scan data, which Neff et al. updated based upon their larger dataset and recently crystal structure and Cryo EM data showing Protoxin-II’s interaction with Nav1.7’s VSD2, and the location of huwentoxin’s binding to VSD2, respectively [Citation51,Citation53,Citation56,Citation58]. A photocrosslinking study by Tzakoniati et al. identified interactions of K27 with E811 and R29 with E759 (and T758) [Citation59]. These results diverge from the Neff et al. model, but the two proposed models agree on the general orientation of HwTX-IV and the interaction of K32 with E811.
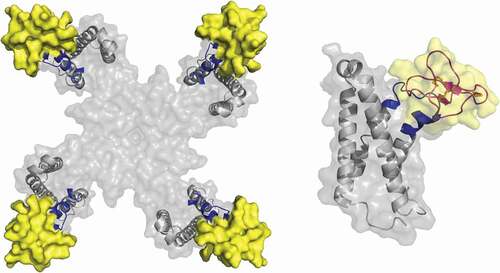
Recently, Guo et al [Citation60] attempted to produce a cryo-EM structure of HwTX-IV bound to a nanodisc-embedded bacterial Nav channel containing the extracellular halves of the human S3-S4 sequence. Although the resolution was insufficient to map side chain orientation, it allowed the docking of HwTX-IV’s NMR structure onto the channel via backbone alignment. This confirmed the general orientation of HwTx-IV in the Neff and Tzakoniati models, and identified I5, F6, and W30 as residues that are immersed in the membrane [Citation60] ().
Figure 1. Left: Cryo-EM structure of huwentoxin-IV (PDB:1MB6, yellow) bound to the chimeric NaChBac (PDB:6 W6O, gray) containing the sequence of the extracellular half of the human VSD2 S3-S4 segments (blue). Right: Side view of huwentoxin docked on the chimeric NaChBac channel
GpTX-1
Scientists at Amgen identified GpTX-1 in the fractionated venom of the Chilean Rose Hair Tarantula (Grammostola portei) by screening fractions of its venom for Nav1.7 activity [Citation61]. This 34-residue peptide contains three disulfide linkages, an amidated C-terminus and mediates its Nav1.7 block through an interaction with VSD2 with an IC50 of 10 nM [Citation61,Citation62]. In patch-clamp experiments synthetically produced GpTX-1 showed a ~ 3-fold improvement in Nav1.7 potency compared to HwTX-IV and a larger selectivity window between Nav1.7 and Nav1.5 activity (>1000-fold vs. 750-fold) but diminished selectivity against Nav1.4 from 120-fold compared to 20-fold [Citation62].
Alanine scanning of GpTX-1 showed that the C-terminus played an integral role in Nav1.7 potency, with H27, W29, K31, K32, and F34 all diminishing potency when mutated. Moderate effects were also observed with alanine substitution at F5 and R7. All of these residues cluster on a single region of the molecule, creating a hydrophobic face that likely mediates critical interactions with sodium channels [Citation62].
Notably, the F5A mutation improved Nav1.4 selectivity to 300-fold. Other small aliphatic substitutions at this position also improved Nav1.4 selectivity while maintaining Nav1.7 potency, whereas the introduction of larger residues reduced Nav1.7 activity. Combinatorial mutation at positions F5 and other locations on the periphery of the hypothesized binding face led to the production of [F5A, M6F, T26L, K28R]-GpTX-1, which had improved Nav1.7 potency, >1000x selectivity against Nav1.4 and Nav1.5, and a mitigated oxidation liability [Citation62].
In addition, a GpTX-1 variant library created through substitution of a subset of amino acids with distinct physical properties at each position in GpTX-1 was also generated [Citation63]. This revealed SAR not predicted by the alanine scan. In addition to confirming the importance of H27, W29, K31, and F34 and the C-terminus in maintaining potency, it also identified positions F5, M6, S24, and Y32 as critical determinants of GpTX-1 activity. Furthermore, this screen identified I10E, P11E, and R18K as substitutions that maintained Nav1.7 potency and selectivity against Nav1.5, and dramatically improved selectivity against Nav1.4 [Citation63].
To extend the half-life of this peptide, various sites away from the putative binding face were PEGylated in [M6F]-GpTX-1. PEG addition at positions D12, N13, K15, and V22 did not disrupt Nav1.7 activity. Identification of these positions facilitated the production of dimeric peptides, which did not produce an avidity effect, but rather maintained potency while rendering the Nav1.7 block essentially irreversible [Citation61]. [F5A, M6F, Pra13, T26L, K28R]-GpTX-1 was also conjugated to a nontargeting anti-2,4-dinitrophenol human IG1 antibody at different conjugation sites on the antibody via a PEG linker of varying lengths [Citation64]. Nav1.7 activity was maintained in conjugates but caused 7- to 230-fold right-shifts in potency relative to GpTX-1, with multivalent molecules being more potent than monovalent ones. Selectivity against Nav1.4 and Nav1.5 also persisted, but was reduced largely due to the Nav1.7 potency decrease [Citation64].
Subcutaneous dosing of the symmetric divalent molecule, E384C-PEG11-[F5A, M6F, Pra13, T26L, K28R]-GpTX-1 resulted in detectable plasma concentrations for 7 days postdosing with minimal proteolytic degradation. Interestingly, the toxin conjugated antibody distributed more effectively to Nav1.7 expressing dorsal root ganglion and sciatic nerve fibers than unconjugated antibodies and accumulated at nerve fibers to a greater extent in wild type compared to Nav1.7 knockout mice. This suggests a role for both Nav.7 and the toxin in this molecule’s distribution [Citation64] – potentially by the toxin–target interaction increasing dwell time at the sensory nerves.
Although the antibody conjugated toxin was able to access nerve fibers, no level of target engagement or efficacy was reported [Citation64]. GpTX-1, however, has been shown to be efficacious in reducing pain behaviors evoked by intraplantar injection of OD-1 [Citation65]. In addition, spinal administration of GpTX-1 and [F5A, M6F, T26L, K28R]-GpTX-1 provided dose-dependent analgesia in mechanical, thermal, visceral, and neuropathic pain models which was undiminished with 8 days of repeated administration [Citation66]. GpTX-1, however, had no effect on OD-1-induced pain behaviors when delivered intraperitoneally at the maximum tolerated dose [Citation65], and no evidence to date has been presented demonstrating efficacy of an engineered peptide in a relevant pain model with systemic GpTX-1 administration.
CcoTx1
Scientists at Pfizer chose ceratotoxin-1 (CcoTX-1), a component of the venom of Ceratogyrus cornuatus, as a starting scaffold for engineering a Nav1.7 selective peptide [Citation67]. CcoTX1 is a potent Nav1.7 and Nav1.2 blocker (IC50 ~ 75 nM) that blocks VGSCs through an interaction with the S3-S4 linker of VSD2, is marginally selective against Nav.16 and is only weakly active against Nav1.4 and Nav1.5 [Citation45,Citation67]. After three rounds of directed evolution variant 2760 ([W5M, K12E, N19R, Y20L, T21V, R25S, D26H, Y31W, D32K]-CcoTX1) was identified. 2760 showed a 3-fold improvement in Nav1.7 potency and a 2-fold decrease in Nav1.2 potency compared to CcoTX-1. 2760 was only 3-fold less potent against Nav1.6 compared to Nav1.7, however [Citation67].
To further improve 2760, the authors performed an amino acid scan of every non-cysteine position and measured activity against VGSCs. This led to the discovery of [D1I]-2760, which improved Nav1.7 potency 2.5-fold, and increased the Nav1.6 and Nav1.2 selectivity to 6- and 16-fold, respectively. Selectivity against Nav1.4 and Nav1.5 was maintained in this peptide [Citation67].
Posttranslational modifications of [D1I]-2760 also led to improvements in this molecule’s activity profile. C-terminal amidation of 2760 and [D1I]-2760 increased Nav1.7 and Nav1.6 potency for both peptides ~5-fold but only increased Nav1.2 potency ~2-fold. In addition, amidated [D1I]-2760 showed broad selectivity against a wide range of ion channels. Introduction of an N terminal pyroglutamate to the amidated [D1I]-2760 produced [D1Za]-2760, which was able to maintain a greater block of Nav1.7 during sustained depolarizations than [D1Ia]-2760 without altering its potency against any of the Nav channels [Citation67].
Solving a crystal structure of CcoTX-1 allowed for the identification of a proposed interaction face comprised of a group of hydrophobic residues (M5, F6, Y28, Y31, L33) and a group of polar or charged residues (R19, H26, K30, K32). Scherbatko et al. then introduced multiple mutations that conferred benefit in their amino acid scanning effort, distant from each other, and that were not a part of the interaction face. These mutants, [D1Z,M5I,K18Y,R24K,a]-2760 and [D1Z,M5I,R27N,a]-2760 both maintained potency for Nav1.7 and improved selectivity for Nav1.2 and Nav1.6 by ~2-fold [Citation67].
As detailed above, a large amount of effort has been directed at engineering Family I scaffolds. Although significant progress has been made in understanding the molecular basis of Nav1.7 inhibition by these peptides, overall, these efforts have not yielded Family 1 related peptides with dramatically improved Nav1.7 selectivity and in-vivo tolerability. Improving the properties of Family 1 peptides is clearly a challenging endeavor. Whether new insights into the structural basis for Nav1.7 inhibition by family I peptides will facilitate the design of new analogs with improved properties remains to be seen, but it seems likely that other families of spider venom peptides might ultimately represent better scaffolds for engineering.
NaSpTx family II
Family 2 is a large family of toxins isolated from tarantula venoms that range in size from 33 to 41 residues and, like family I, they each contain three disulfide bonds that form an ICK motif. Several members of this family have confirmed activity on voltage-gated calcium (CaV) channels and/or voltage-gated potassium (Kv) channels. Only two members of this family, β/ω-TRTX-Tp1a (protoxin I) and μ-TRTX-Pn3a, have potent NaV1.7 blocking activity. To date, engineering efforts on these family II scaffolds have been limited.
Pn3a
Pn3a (μ-TRTX-Pn3a) was isolated from the venom of the South American tarantula Pamphobeteus nigricolor. Characterization using whole-cell electrophysiology in HEK293 cells revealed that Pn3a was a potent inhibitor of NaV1.7 (IC50 0.8 nM), with substantial selectivity other Navs (40-fold over NaV1.1, 100-fold over Nav1.2, Nav1.3 Nav1.4 and Nav1.6 and greater than 900-fold over Nav1.5, Nav1.8 and Nav1.9). Although family II peptides are often regarded as somewhat promiscuous, Pn3a also exhibited substantial selectivity over rat KV2.1, Cav1.2, hCav2.2, α7 nAChR and α3 nAChR, making this peptide one of the most selective Nav1.7 inhibitors reported to date [Citation46]. The analgesic potential of Pn3a was evaluated in multiple rodent models of pain. To assess on-target activity at Nav1.7 in vivo, the ability of Pn3a to inhibit spontaneous pain behaviors induced by intraplantar injection of the Nav1.7 modifying scorpion toxin, OD1 [Citation68] was assessed. Encouragingly, Pn3a (0.3–3 mg/kg IP) inhibited OD-1-induced pain behaviors, confirming on-target Nav1.7 activity in vivo. Despite this apparent on-target activity however, Pn3a displayed no analgesic activity in formalin-, carrageenan- or FCA-induced pain in rodents over the same dose range. The discrepancy between OD-1-induced pain behaviors and other measures of pain is interesting and suggests that maybe lower levels of target engagement are sufficient for activity in the OD-1 model compared to other models. The finding that Pn3a efficacy could be strongly potentiated by subtherapeutic doses of opioids or the enkephalinase inhibitor thiorphan further supports the possibility that Pn3a may exhibit, sub-threshold target occupancy at the doses tested.
In contrast to the findings in the formalin-, carrageenan- and FCA-induced pain models, Pn3a was recently shown to be effective in a mouse model of acute postsurgical pain. In this model, Pn3a completely reversed mechanical hypersensitivity in the absence of adverse motor effects. These findings suggest that different levels of target engagement may be required for efficacy in different pain models [Citation69]. Interestingly, the analgesic effects of Pn3a were completely reversed by co-administration of the opioid receptor antagonist naloxone in this model, suggesting that different levels of endogenous “opiate tone” may contribute to differences in sensitivity to Nav1.7 blockers with sub-threshold levels of target engagement.
The high potency and encouraging selectivity of Pn3a together with evidence of in-vivo target engagement suggests that this family II peptide may represent a good starting point for engineering of Nav1.7 blocking peptides with improved Nav1.7 selectivity and in-vivo tolerability. To date engineering efforts have been limited. Notably, replacement of negatively charged D1 and D8 residues led to improvements in potency, with D8N providing a 20-fold improvement in Pn3a potency at NaV1.7 while maintaining selectivity over other Nav isoforms [Citation70]. Pn3a[D8N] exhibited significantly reduced OD-1-induced pain behaviors and in a model of acute postsurgical mechanical allodynia at doses 3-fold lower than with wild-type Pn3a.
NaSpTx family III
ProTX-II
Family 3 peptides are typically 29–32 residues long, with three disulfide bonds forming an ICK motif. The best characterized member of this family is ProTX-II (β/ω-TRTX-Tp2a). ProTx-II was first isolated from the venom of the Peruvian Green Velvet tarantula, Thrixopelma pruriens, by scientists at Merck based on its ability to inhibit Nav1.8 [Citation47]. Interestingly, the initial characterization also revealed that ProTX-II could potently inhibit Nav1.7 with a significant degree of selectivity over Nav1.5. ProTX-II was subsequently shown to be one of the most potent and selective Nav1.7 blockers known, with an IC50 of 0.3 nM for Nav1.7 and at least 100x fold selectivity over all other voltage-gated sodium-channel isoforms [Citation71]. Although more recent reports suggest that selectivity over key neuronal sodium channels may be lower than first thought (Flinspach et al., 2017; Xu et al., 2019). Like other tarantula toxins, ProTX-II binds to poorly conserved residues in the domain II voltage sensor to inhibit channel activation [Citation56,Citation71,Citation72], a mechanism that has presumably been highly optimized through venom evolution to powerfully inhibit nervous system ion channels under in-vivo conditions.
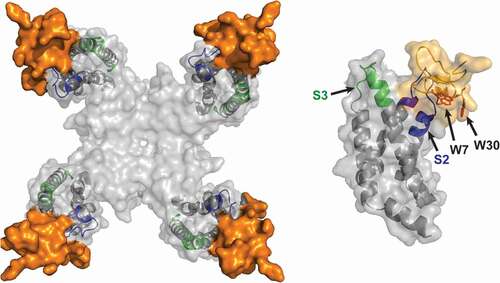
Recently, scientists at Genentech were able to solve the X-ray and cryo-EM structures of ProTX-II in complex with a NavAb construct containing the VSD2 from human Nav1.7 [Citation56]. Their data suggest that the molecular basis of ProTX-II activity is complex, with different structural features involved in lipid binding and orientation, channel interactions, and inhibitory efficacy (). The structure shows that an aromatic-rich surface including W5, W7, W24, and W30 anchors ProTx-II in the membrane, from where it attacks VSD2, perching directly on top of the S3 helix, the most peripherally exposed structural element in Nav channels. A polybasic C-terminal tail (K26-K27-K28) capped by a hydrophobic dyad (L29-W30) further anchors the toxin into the membrane while positioning it over the S3 helix. W24 wedges into a cleft between the lipid facing S2 and S3 helices, while W5 and M6 flank F813, an established determinant for ProTx-II selectivity [Citation71,Citation72]. Two basic ProTX-II side chains, R22 and K26, extend into the extracellular vestibule of VSD2. R22 engages D816 and D818 on the S4 helix, while K26 directly engages E811 on the S3 helix through a sal –bridge interaction, suggesting ProTX-II inhibits Nav1.7 by a simple mechanism involving antagonism of outward gating-charge movement through direct electrostatic repulsion. F813, E811, D816, and E818 are all unique to Nav1.7, suggesting interactions with these residues underlie the selectivity of ProTX-II.
Figure 2. Left: ProTX-II-VSD2-NavAb channel crystal structure (PDB:64NI), top view, with human Nav1.7 VSD2 segments shown in blue (S1–S2) and green (S3–S4). Right: Side view highlighting W7 and W30 on ProTX-II
Binding of ProTX-II to the outer edge of VSD2 was confirmed in a 3.2-Å resolution cryo-EM structure of a ProTX-II bound E406K variant of human Nav1.7 [Citation58]. Unfortunately, the density attributed to ProTX-II was present at peripheral regions and was only resolved to a moderate resolution of ~5 Å, precluding accurate docking of the toxin structures.
Despite impressive potency, coupled with clear evidence of Nav1.7 selectivity and an optimized mechanism of action, the in-vivo analgesic activity of ProTX-II is disappointing. In an initial study, ProTx-II was not efficacious in rodent models of acute and inflammatory pain when delivered by the intravenous route [Citation71]. Lack of analgesic efficacy following systemic administration may reflect an inability of the peptide to cross the blood/nerve barrier (BNB) to reach target sites on sensory nerves [Citation73]. ProTX-II was also initially shown to lack efficacy when delivered by the intrathecal route, which was surprising since access to target sites in the dorsal horn should not be limited when administered by this route [Citation74]. However, it is now clear that ProTX-II can indeed exert strong efficacy following intrathecal administration, but only within a very narrow dose range. Profound motor effects and lethality are observed at doses just above the efficacious dose range making detailed exploration of the dose–response relationship for analgesic activity impossible [Citation75]. Although the exact basis for the narrow therapeutic window for ProTX-II is not fully understood, it seems likely that ProTX-II selectivity is inadequate and inhibition of sodium channel isoforms present on motor neurons, such as Nav1.1 and Nav1.6, underlies the narrow therapeutic window. Thus, although the narrow therapeutic window precludes the use of ProTX-II itself as an analgesic agent, ProTX-II does represent an interesting starting point for the engineering of Nav1.7 blocking peptides with improved Nav1.7 selectivity and in-vivo tolerability.
The first study to explore ProTX-II structure–activity relationships was published by scientists from Purdue Pharma [Citation76]. The aqueous solution structure of ProTx II was solved and indicated that the toxin comprises a well-defined inhibitor cystine knot (ICK) backbone region and a flexible C-terminal tail region. A series of chimeric toxins, amino acid substitutions, and tail region modifications were synthesized and tested for activity against Nav1.7 and Nav1.2. The results of these studies indicated that the flexible tail region plays an important role in the Nav channel potency and to some degree the isoform selectivity of ProTX-II. Strikingly, changing the C-terminal functional group from a carboxylic acid to a methylamide increased the Nav1.7 potency of the resulting compound (compound 17) by 24-fold without changing its selectivity over Nav1.2. These early findings were important for two reasons. Firstly, they showed that the properties of ProTX-II could be modified through sequence modification. Secondly, they identified the flexible C-tail as a potentially fruitful region for further exploration.
To date, the most comprehensive ProTX-II engineering effort was performed by scientists at Janssen R&D. A limited amino acid scan was conducted at all non-cysteine positions using A, D, Q, R, K, and S for diversification [Citation77,Citation78]. From this scan, substitutions Y1Q, W7Q, S11A, were identified that improved potency, selectivity and/or yield of the resulting variants. Combinatorial libraries were designed to test for additive effects of select single position hits to generate Nav1.7 antagonists with further improved potency and selectivity profile compared to the native peptide. This effort led to the identification of several peptides with modestly improved selectivity compared to ProTX-II. NV1D2775-COOH ([Y1Q,W7Q,S11A,M19F]-ProTX-II) was active in the CFA and carrageenan models of inflammatory pain and in the hot plate test, when administered by subcutaneous osmotic mini-pump. NV1D2775-OH also demonstrated significant efficacy in the tail flick, hot plate and formalin tests when administered intrathecally. These results were encouraging, showing for the first time that engineered ProTX-II analogs could exhibit efficacy following systemic dosing. These results also suggested that improvements in in-vitro selectivity might translate directly into improvements in in-vivo tolerability.
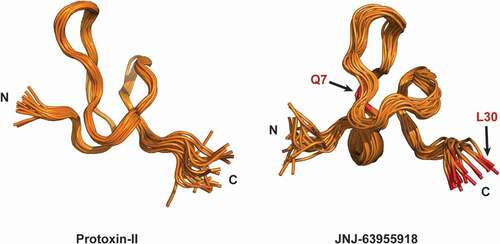
In a second round of engineering, each of the 24 non-cysteine positions in ProTX-II was systematically substituted with every coded amino acid except for methionine and cysteine and single position substitutions that showed improved selectivity or improved recombinant peptide yield were evaluated combinatorially [Citation78]. In total, over 1500 recombinant and synthetic ProTX-II-related peptides were generated. From the initial SAR exploration, hydrophobic substitutions at position 30 were tolerated, with W30L in particular identified as a substitution that significantly improved the sodium channel selectivity in favor of Nav1.7 (IC50 values for ProTX-II W30L were 5 nM, 501 nM, and 4 μM for Nav1.7, Nav1.6, and Nav1.4, respectively). The importance of W30 as a mediator of ProTX-II selectivity is interesting given the key role of this residue for positioning the toxin on top of the S3 helix (). Further improvements in selectivity were observed when W30L was combined with a second substitution, W7Q that additionally improved refolding efficiency (as evidenced by the overall yield from crude in solid-phase peptide synthesis). The resulting peptide, [GP-W7Q, W30L]-ProTX-II or JNJ63955918 exhibited 100- to >1000-fold selectivity over Nav1.1, Nav1.2, Nav1.4, Nav1.5, and Nav1.6. Importantly selectivity over Nav1.1 and Nav1.6 was significantly improved relative to ProTX-II [Citation75]. NMR studies indicated that JNJ63955918 adopts condensed inhibitor cystine knot (ICK) fold stabilized by three conserved disulfide bonds with a flexible C-terminus in solution. The solution structure of JNJ63955918 was very similar to the parent ProTX-II and other related spider venom peptides [Citation75,Citation76,Citation79] (), offering few clues as to the basis of the differential sodium channel activity amongst these family 3 peptides. The impact of the W30L mutation further implicates the flexible C-terminus as a key determinant of sodium channel selectivity. [Citation78]. Other substitutions on the JNJ63955918 scaffold, such as S11V, E17L, and E17N, were well tolerated, with preliminary data suggesting that further gains in potency and/or selectivity might be possible with additional rounds of engineering [Citation78]. A detailed description of these and other mutants has not been published, however.
Figure 3. Side by side comparison of the two structure ensembles of protoxin-II (2N9T) and JNJ63955918 (5TCZ). The global ICK fold is evident in the two highly homologous peptides. 20 lowest energy conformers for each ensemble are shown
Consistent with the improved selectivity, intrathecal JNJ63953918 dose-dependently reduced flinching in phase 1 and phase 2 of the rat formalin model and increased withdrawal latencies in the tail flick and hot plate assays at doses that had no effect on motor function [Citation75]. In side-by-side comparisons, JNJ63955918 was equi-effective to ziconotide in the rat formalin model. However, whereas JNJ63955918 was well tolerated at efficacious doses, ziconotide produced whole body shakes, increased muscle tone and “serpentine” tail movement at all effective doses. Interestingly, peri-sciatic (PS) administration of JNJ63955918 increased the thermal withdrawal latency of the ipsilateral paw in a rat Hargreaves test with no evidence of discoordination or motor effects, indicating that the peptide was also effective by local peripheral administration.
JzTX-V
Scientists at Amgen evaluated venom fractions from the Chinese earth tiger tarantula Chilobrachys jingzhao and identified a 29-residue Family 3 inhibitory cystine knot (ICK) peptide JzTx-V (β/κ-theraphotoxin-Cg2a also known as β-theraphotoxin-Cj2a) with sub-nM NaV1.7 inhibitory activity [Citation80]. In keeping with previous studies (Zeng et al., 2007; Luo et al., 2014), JzTX-V exhibited minimal selectivity over Nav1.4 (3- to 4-fold selectivity). JzTX-V did however, exhibit substantial selectivity over Nav1.5 (~4,000-fold selectivity). Amgen scientists then employed an attribute-based positional scanning strategy [Citation63,Citation81] to improve Nav1.7 selectivity. This effort led to the discovery of a key Ile28Glu mutation that had minimal impact on Nav1.7 potency but imparted >100-fold selectivity for Nav1.7 over Nav1.4, while maintaining substantial selectivity over Nav1.5. These findings represented a significant advance in generating selective Nav1.7 inhibitors from the JzTx-V scaffold. These findings also provided yet another example of a change to the C-terminus of a Family 3 spider toxin having a significant impact on Nav1.7 selectivity. Alanine and glutamic acid scanning mutagenesis of all non-cysteine residues of JzTX-V also identified additional key residues for Nav1.7 block, including Y5, M6, T8, D10, R13, L19, L23, W24, and R26, suggesting that further engineering on the JzTX-V scaffold might be fruitful.
As part of an effort to attach handles for potential derivatization of JzTX-V related peptides, alkyne side-chain containing residues, e.g. propargylglycine (Pra) were incorporated at positions that were expected to have minimal impact on Nav1.7 potency. In addition, Met6 was replaced with the isosteric norleucine, to avoid side-chain oxidation during synthesis and folding. These efforts led to the discovery of AM-8145 (Pra-[Nle6;E28]JzTx-V(1–29)), and AM-0422 (CyA-[Nle6;Pra17;E28]JzTx-V(1–29)) [Citation80]. Both peptides were equipotent with WT JzTx-V, demonstrated excellent selectivity over Nav1.4 and Nav1.5, and exhibited marked selectivity for native Nav1.7 dependent TTX-S currents over native Nav1.7 independent TTX-R currents in mouse DRG neurons. AM-0422 but not a closely related but substantially less active peptide AM-8395 (Pra-[Nle6;E19,28]JzTx-V(1–29)) blocked capsaicin-induced dorsal root ganglion (DRG) neuron action potential firing and mechanically induced C-fiber spiking in a saphenous skin–nerve preparation [Citation80]. Unfortunately, despite a promising in-vitro profile, AM-8145 did not appear to be well tolerated in-vivo (maximally tolerated dose was 1 mg/kg; Cmax ~ 6.5 fold mouse Nav1.7 IC50) and had no effect on Nav1.7-dependent histamine-induced pruritis [Citation82]. These findings suggest that the overall selectivity of AM-8145 was not optimal with inadequate selectivity over Nav1.6 (33 fold) and/or Nav1.1 (47 fold) providing a likely explanation for the poor in-vivo tolerability.
Guided by the single residue scan data described above, extensive further engineering led to the discovery of the potent and selective JzTX-V analog, AM-6120 (E-[Pra1;Nle6;A12;5-BrW24;E28]JzTx-V(1–29)) (IC50 of 0.8 nM); >750-fold selective against Nav1.5, 1.6, and 1.8 [Citation82]. As for other peptides in this series, AM-6120 potently blocked recombinant mouse Nav1.7 and TTX-S currents in mouse DRG, with no effect on TTX-R currents. Importantly, and consistent with improved selectivity, AM-6120 appeared to be better tolerated in-vivo compared to AM-8145, reaching plasma concentrations over 100-fold the in-vitro mouse Nav1.7 IC50 (Cmax was 1.48μM) at a dose that had no effect in a separate open field activity study (2 mg/kg subcutaneous). AM-6120 was efficacious in a Nav1.7-dependent behavioral model of histamine-induced pruritis in mice after subcutaneous administration. The activity was characterized by a steep dose–response relationship reminiscent of the dose–effect relationship for the other Family 3 peptides, ProTX-II and JNJ-63955918 [Citation75].
When considering their potential as systemically acting therapeutics, a major limitation of small peptides is their very short in-vivo half-lives. The fusion of peptides to antibodies, antibody domains (i.e. Fc), or other half-life extending moieties such as PEG or albumin is a potential strategy to maintain the positive attributes of the peptide in a molecule with a long in-vivo half-life. Recently this approach has been successfully applied to create FC and BSA fusion of venom-based peptides targeting Kv1.3 [Citation81,Citation83]. Scientists at Amgen have adopted such an approach to improve the in-vivo half-life of the JzTx-V-based peptides [Citation84]. Architectural variations in the linker, peptide loading, and antibody attachment site were explored to create peptide–antibody conjugates of AM-0422. Although initial attempts were successful at identifying potent conjugates, pharmacokinetic and bioimaging analyses revealed a shorter than expected plasma half-life in vivo with accumulation in the liver. Identifying conjugates with an optimal balance of Nav1.7 potency and half-life required further extensive engineering. Reducing the peptide net charge from +6 to +2 resulted in a conjugate AM-2752, pGlu-[Pra1;Nle6;E12,28;5-BrW24;hF29]JzTx-V(1–29) with maintained Nav1.7 potency along with improved plasma exposure. However, this conjugate exhibited only moderate activity in a Nav1.7-dependent histamine-induced pruritis pharmacodynamic model in mice, suggesting further multiparameter optimization of the peptide, linker, and/or fusion partner is still needed to identify a conjugate that can fully engage NaV1.7 in vivo.
Other family 3 toxins
Numerous other family III toxins including β-TRTX-Gr1a (GrTx1), β-TRTX-Gr1b (GsAF I), β/κ-TRTX-Pm2a (Pterinotoxin-2), κ-TRTX-Gr2a (GsMTx-2), κ-TRTX-Ps1a (Phrixotoxin-1), are known to possess potent Nav1.7 activity [Citation56], but selectivity and suitability of these peptides as scaffolds for engineering have not been investigated in detail.
Outlook
Tremendous progress in peptide engineering has been made over the last decade, particularly with Family 3 scaffolds, culminating in the identification of JNJ-63955918 and AM-6120 as potent, highly selective closed-state Nav1.7 blockers that exert convincing efficacy at well-tolerated doses in-vivo. These discoveries represent a significant step forward in the search for Nav1.7-based analgesics and local delivery of JNJ63955918, AM-6120, or related peptides, which could provide a viable strategy for the treatment of certain forms of severe pain. The recent elucidation of the structural basis for Nav1.7 inhibition by ProTX-II should facilitate the design of ProTX-II analogs with even greater selectivity and in-vivo tolerability. Furthermore, additional peptides from families I and III, or from other families of spider venoms have not been explored and may provide significant new opportunities for engineering selective Nav1.7 blockers.
Identification of peptide-based Nav1.7 blockers suitable for long-term systemic use, however, continues to be challenging. A much deeper understanding of the factors that determine efficacy and safety, such as pharmacokinetics, site of action (axonal, DRG, or central nerve terminals) and tissue distribution, levels of target engagement, immunogenicity and other factors such as plasma protein binding, efflux, and metabolism will be required before peptides can be suitably optimized for long-term use. Clearly, much remains to be done but the encouraging progress highlighted in this article suggests that engineered spider venoms represent a valid approach for targeting Nav1.7 that might finally allow the translation of the promise of Nav1.7 into clinical success for patients in desperate need of new analgesic options.
Disclosure statement
The authors are employees of Janssen R&D, LLC.
References
- Hubner CA, Jentsch TJ. Ion channel diseases. Hum Mol Genet. 2002;11:2435–2445.
- Yogeeswari P, Ragavendran JV, Thirumurugan R, et al. Ion channels as important targets for antiepileptic drug design. Curr Drug Targets. 2004;5:589–602.
- Tfelt-Hansen J, Winkel BG, Grunnet M, et al. Inherited cardiac diseases caused by mutations in the Nav1.5 sodium channel. J Cardiovasc Electrophysiol. 2010;21:107–115.
- Cannon SC, Bean BP. Sodium channels gone wild: resurgent current from neuronal and muscle channelopathies. J Clin Invest. 2010;120:80–83.
- Cregg R, Momin A, Rugiero F, et al. Pain channelopathies. J Physiol. 2010;588:1897–1904.
- Toledo-Aral JJ, Moss BL, He Z-J, et al. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A. 1997;94:1527–1532.
- Alexandrou AJ, Brown AR, Chapman ML, et al. Subtype-selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PloS One. 2016;11:e0152405.
- Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41:171–174.
- Fertleman CR, Baker MD, Parker KA, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774.
- Legroux-Crespel E, Sassolas B, Guillet G, et al. [Treatment of familial erythermalgia with the association of lidocaine and mexiletine]. Annales de dermatologie et de venereologie. 2003;130:429–433.
- Li QS, Cheng P, Favis R, et al. SCN9A variants may be implicated in neuropathic pain associated with diabetic peripheral neuropathy and pain severity. Clin J Pain. 2015;31:976–982.
- Reimann F, Cox JJ, Belfer I, et al. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc Natl Acad Sci U S A. 2010;107:5148–5153.
- Estacion M, Harty TP, Choi JS, et al. A sodium channel gene SCN9A polymorphism that increases nociceptor excitability. Ann Neurol. 2009;66:862–866.
- Vargas-Alarcon G, Alvarez-Leon E, Fragoso J-M, et al. A SCN9A gene-encoded dorsal root ganglia sodium channel polymorphism associated with severe fibromyalgia. BMC Musculoskelet Disord. 2012;13:23.
- Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898.
- Goldberg YP, MacFarlane J, MacDonald ML, et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet. 2007;71:311–319.
- Sun J, Li L, Yang L, et al. Novel SCN9A missense mutations contribute to congenital insensitivity to pain: unexpected correlation between electrophysiological characterization and clinical phenotype. Mol Pain. 2020;16:1744806920923881.
- He W, Young GT, Zhang B, et al. Functional confirmation that the R1488* variant in SCN9A results in complete loss-of-function of Nav1.7. BMC Med Genet. 2018;19:124.
- Cox JJ, Sheynin J, Shorer Z, et al. Congenital insensitivity to pain: novel SCN9A missense and in-frame deletion mutations. Hum Mutat. 2010;31:E1670–1686.
- Kurban M, Wajid M, Shimomura Y, et al. A nonsense mutation in the SCN9A gene in congenital insensitivity to pain. Dermatology. 2010;221:179–183.
- Nilsen KB, Nicholas AK, Woods CG, et al. Two novel SCN9A mutations causing insensitivity to pain. Pain. 2009;143:155–158.
- Shorer Z, Wajsbrot E, Liran TH, et al. A novel mutation in SCN9A in a child with congenital insensitivity to pain. Pediatr Neurol. 2014;50:73–76.
- Wheeler DW, Lee MC, Harrison EK, et al. Case Report: neuropathic pain in a patient with congenital insensitivity to pain. F1000Res. 2014;3:135.
- Ahmad S, Dahllund L, Eriksson AB, et al. A stop codon mutation in SCN9A causes lack of pain sensation. Hum Mol Genet. 2007;16:2114–2121.
- Bogdanova-Mihaylova P, Alexander MD, Murphy RP, et al. SCN9A-associated congenital insensitivity to pain and anosmia in an Irish patient. J Peripher Nerv Syst: JPNS. 2015;20:86–87.
- Marchi M, Provitera V, Nolano M, et al. A novel SCN9A splicing mutation in a compound heterozygous girl with congenital insensitivity to pain, hyposmia and hypogeusia. J Peripher Nerv Syst: JPNS. 2018;23:202–206.
- Staud R, Price DD, Janicke D, et al. Two novel mutations of SCN9A (Nav1.7) are associated with partial congenital insensitivity to pain. Eur J Pain. 2011;15:223–230.
- Weiss J, Pyrski M, Jacobi E, et al. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature. 2011;472:186–190.
- Bartholomew F, Lazar J, Marqueling A, et al. Channelopathy: a novel mutation in the SCN9A gene causes insensitivity to pain and autonomic dysregulation. Br J Dermatol. 2014;171:1268–1270.
- Yuan J, Matsuura E, Higuchi Y, et al. Hereditary sensory and autonomic neuropathy type IID caused by an SCN9A mutation. Neurology. 2013;80:1641–1649.
- Gingras J, Smith S, Matson DJ, et al. Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PloS One. 2014;9:e105895.
- Nassar MA, Stirling LC, Forlani G, et al. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A. 2004;101:12706–12711.
- Minett MS, Nassar MA, Clark AK, et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun. 2012;3:791.
- Mulcahy JV, Pajouhesh H, Beckley JT, et al. Challenges and Opportunities for Therapeutics Targeting the Voltage-Gated Sodium Channel Isoform NaV1.7. J Med Chem. 2019;62:8695–8710.
- Lee JH, Park C-K, Chen G, et al. A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell. 2014;157:1393–1404.
- Robinson SD, Undheim EAB, Ueberheide B, et al. Venom peptides as therapeutics: advances, challenges and the future of venom-peptide discovery. Expert Rev Proteomics. 2017;14:931–939.
- Dongol Y, Cardoso FC, Lewis RJ. Spider knottin pharmacology at voltage-gated sodium channels and their potential to modulate pain pathways. Toxins (Basel). 2019;11. DOI:https://doi.org/10.3390/toxins11110626.
- Langenegger N, Nentwig W, Kuhn-Nentwig L. Spider venom: components, modes of action, and novel strategies in transcriptomic and proteomic analyses. Toxins (Basel). 2019;11. DOI:https://doi.org/10.3390/toxins11100611.
- Wu T, Wang M, Wu W, et al. Spider venom peptides as potential drug candidates due to their anticancer and antinociceptive activities. J Venom Anim Toxins Incl Trop Dis. 2019;25:e146318.
- Goncalves TC, Benoit E, Partiseti M, et al. The NaV1.7 channel subtype as an antinociceptive target for spider toxins in adult dorsal root ganglia neurons. Front Pharmacol. 2018;9:1000.
- Saez NJ, Senff S, Jensen JE, et al. Spider-venom peptides as therapeutics. Toxins (Basel). 2010;2:2851–2871.
- Klint JK, Senff S, Rupasinghe DB, et al. Spider-venom peptides that target voltage-gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon: Offl J Int Soc Toxinol. 2012;60:478–491.
- Cardoso FC, Lewis RJ. Structure-function and therapeutic potential of spider venom-derived cysteine knot peptides targeting sodium channels. Front Pharmacol. 2019;10:366.
- Peng K, Shu Q, Liu Z, et al. Function and solution structure of huwentoxin-IV, a potent neuronal tetrodotoxin (TTX)-sensitive sodium channel antagonist from Chinese bird spider Selenocosmia huwena. J Biol Chem. 2002;277:47564–47571.
- Bosmans F, Rash L, Zhu S, et al. Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol Pharmacol. 2006;69:419–429.
- Deuis JR, Dekan Z, Wingerd JS, et al. Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci Rep. 2017;7:40883.
- Middleton RE, Warren VA, Kraus RL, et al. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry. 2002;41:14734–14747.
- NCBI:txid29017, NCBI Taxonomy Browser, 1 Dec. 2020, <https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi>
- Xiao Y, Bingham J-P, Zhu W, et al. Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. J Biol Chem. 2008;283:27300–27313.
- Revell JD, Lund P-E, Linley JE, et al. Potency optimization of Huwentoxin-IV on hNav1.7: a neurotoxin TTX-S sodium-channel antagonist from the venom of the Chinese bird-eating spider Selenocosmia huwena. Peptides. 2013;44:40–46.
- Minassian NA, Gibbs A, Shih AY, et al. Analysis of the structural and molecular basis of voltage-sensitive sodium channel inhibition by the spider toxin huwentoxin-IV (mu-TRTX-Hh2a. J Biol Chem. 2013;288:22707–22720.
- Rahnama S, Deuis JR, Cardoso FC, et al. The structure, dynamics and selectivity profile of a NaV1.7 potency-optimised huwentoxin-IV variant. PloS One. 2017;12:e0173551.
- Neff RA, Flinspach M, Gibbs A, et al. Comprehensive engineering of the tarantula venom peptide huwentoxin-IV to inhibit the human voltage-gated sodium channel hNav1.7. J Biol Chem. 2020;295:1315–1327.
- Deng M, Luo X, Jiang L, et al. Synthesis and biological characterization of synthetic analogs of Huwentoxin-IV (Mu-theraphotoxin-Hh2a), a neuronal tetrodotoxin-sensitive sodium channel inhibitor. Toxicon: Offl J Int Soc Toxinol. 2013;71:57–65.
- Agwa AJ, Lawrence N, Deplazes E, et al. Spider peptide toxin HwTx-IV engineered to bind to lipid membranes has an increased inhibitory potency at human voltage-gated sodium channel hNaV1.7. Biochim Biophys Acta. 2017;1859:835–844.
- Xu H, Li T, Rohou A, et al. Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Cell. 2019;176:1238–1239.
- Agwa AJ, Tran P, Mueller A, et al. Manipulation of a spider peptide toxin alters its affinity for lipid bilayers and potency and selectivity for voltage-gated sodium channel subtype 1.7. J Biol Chem. 2020;295:5067–5080.
- Shen H, Liu D, Wu K, et al. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science. 2019;363:1303–1308.
- Tzakoniati F, Xu H, Li T, et al. Development of photocrosslinking probes based on huwentoxin-IV to map the site of interaction on Nav1.7. Cell Chem Biol. 2020;27:306–313 e304. .
- Gao S, Valinsky WC, On NC, et al. Employing NaChBac for cryo-EM analysis of toxin action on voltage-gated Na(+) channels in nanodisc. Proc Natl Acad Sci U S A. 2020;117:14187–14193.
- Murray JK, Biswas K, Holder JR, et al. Sustained inhibition of the NaV1.7 sodium channel by engineered dimers of the domain II binding peptide GpTx-1. Bioorg Med Chem Lett. 2015;25:4866–4871.
- Murray JK, Ligutti J, Liu D, et al. Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the Na(V)1.7 sodium channel. J Med Chem. 2015;58:2299–2314.
- Murray JK, Long J, Zou A, et al. Single Residue Substitutions That Confer Voltage-Gated Sodium Ion Channel Subtype Selectivity in the NaV1.7 Inhibitory Peptide GpTx-1. J Med Chem. 2016;59:2704–2717.
- Biswas K, Nixey TE, Murray JK, et al. Engineering antibody reactivity for efficient derivatization to generate NaV1.7 Inhibitory GpTx-1 peptide-antibody conjugates. ACS Chem Biol. 2017;12:2427–2435.
- Deuis JR, Wingerd J, Winter Z, et al. Analgesic Effects of GpTx-1, PF-04856264 and CNV1014802 in a Mouse Model of NaV1.7-Mediated Pain. Toxins (Basel). 2016;8. DOI:https://doi.org/10.3390/toxins8030078.
- Chen C, Xu B, Shi X, et al. GpTx-1 and [Ala(5), Phe(6), Leu(26), Arg(28)]GpTx-1, two peptide NaV 1.7 inhibitors: analgesic and tolerance properties at the spinal level. Br J Pharmacol. 2018;175:3911–3927.
- Shcherbatko A, Rossi A, Foletti D, et al. Engineering Highly Potent and Selective Microproteins against Nav1.7 Sodium Channel for Treatment of Pain. J Biol Chem. 2016;291:13974–13986.
- Jalali A, Bosmans F, Amininasab M, et al. OD1, the first toxin isolated from the venom of the scorpion Odonthobuthus doriae active on voltage-gated Na+ channels. FEBS Lett. 2005;579:4181–4186.
- Mueller A, Starobova H, Morgan M, et al. Antiallodynic effects of the selective NaV1.7 inhibitor Pn3a in a mouse model of acute postsurgical pain: evidence for analgesic synergy with opioids and baclofen. Pain. 2019;160:1766–1780.
- Mueller A, Dekan Z, Kaas Q, et al. Mapping the Molecular Surface of the Analgesic NaV1.7-Selective Peptide Pn3a Reveals Residues Essential for Membrane and Channel Interactions. ACS Pharmacol Transl Sci. 2020;3:535–546.
- Schmalhofer WA, Calhoun J, Burrows R, et al. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol Pharmacol. 2008;74:1476–1484.
- Xiao Y, Blumenthal K, Jackson JO 2nd, et al. The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol Pharmacol. 2010;78:1124–1134.
- Hackel D, Krug SM, Sauer R-S, et al. Transient opening of the perineurial barrier for analgesic drug delivery. Proc Natl Acad Sci U S A. 2012;109:E2018–2027.
- Black JA, Frezel N, Dib-Hajj SD, et al. Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain. 2012;8:82.
- Flinspach M, Xu Q, Piekarz AD, et al. Insensitivity to pain induced by a potent selective closed-state Nav1.7 inhibitor. Sci Rep. 2017;7:39662.
- Park JH, Carlin KP, Wu G, et al. Studies examining the relationship between the chemical structure of protoxin II and its activity on voltage gated sodium channels. J Med Chem. 2014;57:6623–6631.
- Flinspach M, Neff R, Liu Y, et al. Protoxin-II variants and methods of use. US20150099705. 2015.
- Flinspach M, Fellows R, Xu Q, et al. Protoxin-II Variants and Methods of Use. 2016.
- Chagot B, Escoubas P, Villegas E, et al. Solution structure of Phrixotoxin 1, a specific peptide inhibitor of Kv4 potassium channels from the venom of the theraphosid spider Phrixotrichus auratus. Protein Sci. 2004;13:1197–1208.
- Moyer BD, Murray JK, Ligutti J, et al. Pharmacological characterization of potent and selective NaV1.7 inhibitors engineered from Chilobrachys jingzhao tarantula venom peptide JzTx-V. PloS One. 2018;13:e0196791.
- Murray JK, Qian Y-X, Liu B, et al. Pharmaceutical optimization of peptide toxins for ion channel targets: potent, selective, and long-lived antagonists of Kv1.3. J Med Chem. 2015;58:6784–6802.
- Wu B, Murray JK, Andrews KL, et al. Discovery of tarantula venom-derived NaV1.7-Inhibitory JzTx-V Peptide 5-Br-Trp24 Analogue AM-6120 with Systemic Block of Histamine-Induced Pruritis. J Med Chem. 2018;61:9500–9512.
- Edwards W, Fung-Leung W-P, Huang C, et al. Targeting the ion channel Kv1.3 with scorpion venom peptides engineered for potency, selectivity, and half-life. J Biol Chem. 2014;289:22704–22714.
- Murray JK, Wu B, Tegley CM, et al. Engineering NaV1.7 Inhibitory JzTx-V Peptides with a potency and basicity profile suitable for antibody conjugation to enhance pharmacokinetics. ACS Chem Biol. 2019;14:806–818.