
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The voltage-gated sodium channel Nav1.8 mediates the tetrodotoxin-resistant (TTX-R) Na+ current in nociceptive primary sensory neurons, which has an important role in the transmission of painful stimuli. Here, we describe the functional modulation of the human Nav1.8 α-subunit in Xenopus oocytes by auxiliary β subunits. We found that the β3 subunit down-regulated the maximal Na+ current amplitude and decelerated recovery from inactivation of hNav1.8, whereas the β1 and β2 subunits had no such effects. The specific regulation of Nav1.8 by the β3 subunit constitutes a potential novel regulatory mechanism of the TTX-R Na+ current in primary sensory neurons with potential implications in chronic pain states. In particular, neuropathic pain states are characterized by a down-regulation of Nav1.8 accompanied by increased expression of the β3 subunit. Our results suggest that these two phenomena may be correlated, and that increased levels of the β3 subunit may directly contribute to the down-regulation of Nav1.8. To determine which domain of the β3 subunit is responsible for the specific regulation of hNav1.8, we created chimeras of the β1 and β3 subunits and co-expressed them with the hNav1.8 α-subunit in Xenopus oocytes. The intracellular domain of the β3 subunit was shown to be responsible for the down-regulation of maximal Nav1.8 current amplitudes. In contrast, the extracellular domain mediated the effect of the β3 subunit on hNav1.8 recovery kinetics.
Introduction
Voltage-gated sodium channels (VGSCs), which mediate the rising phase of the action potential in excitable cells, consist of a 260 kD pore-forming α subunit of which there are nine mammalian subtypes known (Nav1.1-Nav1.9), each with distinct tissue distribution and biophysical properties. The α subunits associate with one or more auxiliary β subunits of which there are four known subtypes: β1 (36 kD), β2 (33 kD) [Citation1,Citation2], β3 [Citation3] and β4 (38 kD) [Citation4]. The α subunits consist of four domains, each containing six transmembrane helices flanked by intracellular N- and C-termini; whereas the β subunits all adopt the immunoglobulin-like fold with an intracellular C-terminus, one α-helical membrane-spanning domain and two extracellular β-sheets [Citation5–7]. Although the α subunit alone is sufficient for the formation of a functional channel pore, the β subunits are required for the physiological kinetics and voltage-dependent gating observed in native cells [Citation8].
Primary sensory neurons (dorsal root, nodose and trigeminal ganglion neurons) express a multitude of VGSC α subunit subtypes [Citation9–13] including Nav1.1, 1.2, 1.6, 1.7, 1.8 and 1.9 as well as all four β subunits [Citation3,Citation4,Citation14]. Chronic pain states of both inflammatory and neuropathic origin are characterized by changes in the expression profile of VGSCs in sensory neurons, which in turn leads to altered neuronal excitability. In particular, the VGSC subtype Nav1.8 plays a major role in pain, as demonstrated in Nav1.8 knockout mice, which display attenuated pain behavior in comparison to wild-type mice [Citation15]. Nav1.8 is now considered to have key roles in both inflammatory and neuropathic pain [Citation16], and gain-of-function point mutations in Nav1.8 have been reported in humans with painful neuropathy [Citation17]. Furthermore, Nav1.8 is thought to be the most important VGSC for low temperature-induced pain [Citation18]. Inhibition of Nav1.8 can reduce inflammatory and/or neuropathic pain in animal models [Citation19], demonstrating a key role for this VGSC α subunit in pain states. Nav1.8 is selectively expressed by small dorsal root ganglion (DRG) neurons involved in nociception [Citation12], and mediates a slowly-inactivating tetrodotoxin-resistant (TTX-R) Na+ current which is up-regulated in inflammatory pain states. In contrast, Nav1.8 is down-regulated in neuropathic pain but still considered important in influencing neuronal excitability [Citation20–24]. These changes are partially attributed to alterations in the levels of growth factors that regulate channel expression, however, other mechanisms, such as altered modulation by auxiliary β subunits, may also be involved [Citation25,Citation26]. β subunits can significantly modulate the properties of VGSC α subunits by regulating kinetics and voltage-dependence of gating, regulating cell surface expression levels and act as adhesion molecules (see review [Citation16]). In both rat [Citation5,Citation27] and human DRG [Citation28], neuropathic pain is characterized by an increase in immunoreactivity for the β3 subunit. The β3 subunit co-localizes with Nav1.8 in sensory neurons [Citation29], suggesting that an up-regulation of the β3 subunit may affect the activity and biophysical properties of Nav1.8.
The expression of rat Nav1.8 (rNav1.8) in Xenopus oocytes [Citation5,Citation30] and mammalian cells [Citation31–33] has been described previously, and modulation of rNav1.8 by auxiliary β subunits has been characterized [Citation5,Citation30]. The expression of human Nav1.8 (hNav1.8) in Xenopus oocytes [Citation34], human embryonic kidney (HEK293) cells [Citation35], and mammalian sensory neuron-derived ND7/23 cells [Citation36] has also been reported previously. Although some aspects of β subunit modulation of hNav1.8 have been described previously in mammalian cells [Citation37], others remain to be characterized, such as β subunit-mediated effects on recovery from inactivation. A previous study examined how β1 affected repriming of rNav1.8 [Citation30] but the functional modulation of hNav1.8 by β subunits expressed in Xenopus oocytes has not been characterized. The Xenopus oocyte expression system is useful for screening ion channel targeting compounds [Citation38]. When screening for compounds that can inhibit or modulate hNav1.8, this α subunit should ideally be expressed together with β subunits to better mimic the in vivo situation. Thus, it is important to evaluate how β subunits modulate hNav1.8 in the Xenopus oocyte expression system.
In the present study, we describe the modulation of hNav1.8 by auxiliary β subunits in Xenopus oocytes. We found that β3 affected the maximal current amplitude and recovery from inactivation whereas the β1 and β2 subunits had little influence on these parameters. Both the extracellular [Citation4,Citation39] and intracellular domains of β subunits can interact with the VGSC α subunit [Citation40,Citation41]. To investigate which domains of the β3 subunit mediated these specific effects, we also studied the modulation of hNav1.8 current amplitude and repriming by chimeric β1/β3 and β3/β1subunits.
Materials and methods
cRNA preparation
Rat β1 and β2 subunits were gifts (Dr A.L. Goldin, UC Irvine, CA). Rat β3 cDNA was cloned by RT-PCR, subcloned into the pNKS2 oocyte expression vector [Citation42] and C-terminally fused to a hexahistidine tag. Constructs encoding human Nav1.8 (cloned into pcDNA3.1), rat Nav1.2 (cloned into pLCT1) rat β1, rat β2, rat β3, rat β3[L8F, R20S, F174L,V210A] as well as rat β1/β3 and β3/β1 chimeras (all in pNKS2) were linearized and cRNA was synthesized using SP6 or T7 in vitro transcription kits (Ambion mMessage mMachine, Austin, TX) as described previously [Citation34].
Oocyte preparation and microinjection
Xenopus oocytes were defolliculated with collagenase (Type I, Sigma) at 3 mg/ml in OR-2 medium that contained (mM): 82.5 NaCl, 2 KCl, 1 MgCl2, 5 HEPES-NaOH, pH 7.4 for 2–3 hours at room temperature. Oocytes were stored at 18°C in sterile ND96 medium containing (mM): 96 NaCl, 2 KCl, 1.8 CaCl2, 5 HEPES-NaOH, pH 7.4 supplemented with 5 mM pyruvate and 50 µg/ml gentamycin. Glass pipettes for microinjection were pulled from glass capillaries (3–000-203 GX, Drummond Scientific Co., Broomall, PA). The cRNAs were diluted in water to 0.5 μg/μl, and then diluted further to the appropriate concentrations to inject a total of 2.5 ng of RNA for the hNav1.8 α subunit, alone or in combination with 0.5–5 ng RNA for the β subunits as outlined for each experiment. 50 nL RNA was injected into each oocyte using a microinjector (Nanojet II, Drummond Scientific Co.).
Analysis of expression and glycosylation status of the β3 subunit
Xenopus laevis oocytes were injected with 50 nl aliquots of β3 cRNA (0.5 mg/ml). For metabolic labeling of total protein, oocytes were incubated overnight at 19°C with L-[35S]-methionine at ~100 Mbq/ml with ~ 0.2 MBq/oocyte (Amersham) in sterile ND96 (96 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2 and 5 mM HEPES, pH 7.4). For selective labeling of β3 protein at the plasma membrane, oocytes were cultured for three days after cRNA injection. Intact oocytes were then treated with [125I]-sulfo-SHPP (Amersham), a membrane impermeable derivative of the Bolton-Hunter reagent. Sulfo-SHPP (Pierce) was radioiodinated as described previously [Citation43]. At ambient temperature the following reagents were rapidly and subsequently added to 0.5 μg sulfo-SHPP in 2 μl DMSO: 18.5 MBq of carrier-free Na125I, 10 μl 0.5% chloramine T in 0.5 M sodium phosphate buffer pH 7.5, 100 μl 0.1% DL-α-hydroxyphenyl acetic acid in 0.1 M NaCl, and 10 μl 1.2% sodium metabisulfite in 0.05 M sodium phosphate buffer pH 7.5. 30 μl aliquots of this mixture were immediately added per 10–12 oocytes. After 60 min incubation on ice with occasional gentle mixing, oocytes were washed in ND96 and His-tagged protein was purified via Ni2+-NTA agarose beads (Qiagen) as described previously [Citation43] using 0.5% n-dodecyl-β-D-maltoside (ULTROL Grade, Calbiochem-Novabiochem GmbH, Bad Soden, Germany) as detergent. Shortly, oocytes were homogenized in 0.1 M phosphate buffer (20 μl per oocyte) containing 0.4 mM Pefabloc® SC (Fluka, Buchs, Switzerland) and 0.5% n-dodecyl-β-D-maltoside (ULTROL Grade, Calbiochem-Novabiochem GmbH). The homogenate was incubated on ice for 15 min and the extract was then cleared by centrifugation (10 min at 15,000 rpm in a desktop centrifuge). 100 μl of the clear supernatant were diluted with 400 μl of the above buffer and supplemented with 30 μl Ni2+-NTA agarose beads and 10 mM imidazole. After 30 min of incubation under continuous inversion, the agarose-bound protein was washed four times with 1 ml phosphate buffer containing 0.1% dodecyl maltoside, 0.4 mM Pefabloc® SC, and 25 mM imidazole. Subsequently, protein was eluted from the agarose beads with non-denaturing elution buffer (20 mM Tris-HCl, 100 mM imidazole-HCl, 10 mM EDTA and 0.5% dodecyl maltoside, pH 7.8). Purified protein was kept at 0°C until analyzed. 10 μl aliquots of protein were supplemented with SDS sample buffer and separated on 10% polyacrylamide gels. Gels were dried and exposed to BioMax MR films (Kodak) at – 80°C. For analysis of the glycosylation status, 10 ml aliquots of purified protein were supplemented with reducing (20 mM DTT) SDS sample buffer and 1% octylglucoside (Calbiochem-Novabiochem GmbH) and incubated for 1 h at 37°C with 0.5 or 5 IUB milliunits endoglycosidase H (Endo H) or 5 IUB milliunits PNGase F (New England Biolabs GmbH, Frankfurt, Germany).
Construction of the β subunit chimeras
A Bsm I site was introduced at position 654 of the rat β3 sequence (in pNKS2) using site-directed mutagenesis (QuikChange® II XL, Stratagene, CA). Rat β1 (with naturally occurring Bsm I site at position 805) and rat β3 pNKS2 constructs were digested with Bsm I and either 5ʹ or 3ʹ vector restriction sites to excise the intracellular or extracellular β subunit fragments respectively. Digested fragments were gel purified using a QIAquick gel extraction kit (QIAGEN, Germany) and ligated with T4 DNA Ligase (Fermentas) to obtain the β1/β3 and β3/β1 chimeras. Chimera junctions were checked for correct β subunit switching by DNA sequencing.
Electrophysiological Recording of Na+ currents
Whole cell depolarization-activated currents mediated by hNav1.8 or rNav1.2 were recorded from Xenopus oocytes 3 days after cRNA injection using the two-electrode (virtual ground circuit) voltage clamp technique. Oocytes were placed in a (~400 µl) bath containing the appropriate recording solution mounted on the stage of a dissecting microscope, impaled with glass electrodes and voltage-clamped using a GeneClamp 500B amplifier (Axon Instruments) or an OpusXpress work station (Molecular Devices, Union City, CA). Microelectrodes were pulled from borosilicate glass (GC150TF, Harvard Apparatus) and typically had resistances of 0.3–1.5 MΩ when filled with 3 M KCl. All recordings were made at room temperature (20–23°C). During recordings, oocytes were perfused continuously at a rate of ~1.5 ml/min. Voltage-steps were generated using pCLAMP8 or OpusXpress software (Molecular Devices). Data were low pass filtered at 1 kHz, digitized at 10 kHz, and leak-subtracted on-line using a – P/6 protocol and analyzed off-line. Tetrodotoxin (TTX) was applied via a gravity-fed perfusion system. For each experiment, at least 3 different batches of oocytes were used.
Data analysis
All data were analyzed using Clampfit 8 software (Molecular Devices) and graphs and curves were constructed and analyzed using GraphPad Prism 4.0 (San Diego, CA). Mathematical formulas are described below. All statistical analyses was performed using one-way ANOVA with Tukey’s multiple comparison test, or, when indicated in the figure legend, two-tailed T-test.
The voltage-dependence of activation was determined by measuring the amplitude of the Na+ current elicited by depolarization to various membrane potentials. Voltage-dependent Na+ conductance (G) was determined from transformations of current-voltage relationship (I–V) curves using the formula:
where I is peak current amplitude, V is the test membrane potential, and Vr is the measured or extrapolated reversal potential. Current activation curves were fitted with a sigmoidal Boltzmann function that identifies the voltage at which the VGSC is half-maximally activated:
where G represents the conductance at various membrane potentials, G0 is peak conductance, V0.5 is the voltage where the VGSCs are half-maximally activated, V is the depolarized membrane potential and Kv is the slope constant.
Steady-state inactivation at various membrane potentials was determined by applying 1s pre-pulses to different voltages ranging from – 120 mV to 0 mV immediately followed by a test pulse to the membrane potential generating peak Na+ current. The Na+ current amplitude elicited by the test pulse was normalized to the amplitude elicited after a pre-pulse to – 120 mV, where steady-state inactivation is minimal. Inactivation curves were fitted with a single Boltzmann function:
where I/I0 represents the fraction of current available, V0.5 is the voltage where the VGSCs are half-maximal inactivated, V is the depolarized membrane potential and Kv is the slope constant.
To determine recovery from inactivation of VGSCs, Xenopus oocytes were depolarized to 0 mV for 1 s to inactivate VGSCs and allowed different time periods (2.5 ms – 1 s) to recover at – 70 mV before a depolarizing pulse was applied to generate peak Na+ current. The Na+ current elicited by this pulse (I) was normalized to the current amplitude of an identical pulse not preceded by an inactivating pulse (I0). The fraction of Na+ current recovered was plotted against recovery time and fitted with single or multiple exponential equations of the form:
where I/I0 represents the fraction of recovered current; t represents the recovery time; F1 and F2 represent the fractions of current recovering with the time constants τ1 and τ2.
The time constants for current inactivation were determined by fitting a single or double exponential function to the decay phase of the current:
where I/I0 represents the fraction of current remaining, t represents time and τ1 (and τ2) the time constant (s) for inactivation. F1 and F2 represent the fraction of current in phase when the decay curve was fitted with a double exponential function. In contrast to the other curves which were analyzed by GraphPad Prism 4.0, this analysis was performed directly in ClampFit 8.
Results
Functional expression of human of Nav1.2 and Nav1.8 in Xenopus oocytes
When expressed in Xenopus oocytes, hNav1.8 produced a slowly-inactivating depolarization-activated Na+ current, which was not affected by 1 µM TTX (), as has been shown previously [Citation33]. The TTX-S VGSC subtype rNav1.2 was expressed for comparison. In contrast to hNav1.8, the Na+ current mediated by rNav1.2 exhibited fast activation and inactivation kinetics and was completely blocked by 1 µM TTX (). In the absence of β subunits, the voltage-dependence of activation and inactivation were both best fitted with single Boltzmann functions. Half-maximal activation (V0.5) was determined to be – 2.9 ± 0.4 mV (n = 56), whereas half-maximal inactivation occurred at – 43.5 ± 0.7 mV (n = 40) (). Recovery from inactivation consisted of two phases with distinct time constants ().
Table 1. Effects of the β1, β2 and β3 subunits on biophysical properties of hNav1.8
Biochemical confirmation of synthesis and surface expression of the β3 subunit
The rat β1 and β2 subunits had previously been shown to express and functionally modulate VGSCs in Xenopus oocytes [Citation44]. To demonstrate that the β3 subunit was expressed and localized to the plasma membrane, oocytes were injected with a cRNA encoding a histidine (His)-tagged β3 subunit (β3-His) and metabolically labeled total β3-His protein as well as the selectively radio-iodinated membrane fraction of β3-His were purified via Ni-NTA agarose and analyzed by SDS-PAGE with and without prior endoglycosidase treatment. The β3 subunit was efficiently expressed in the plasma membrane and showed a uniform band of complex glycosylated protein, even in the absence of an α-subunit (). Deglycosylation with PNGase revealed the predicted size of ~30 kDa whereas partial deglycosylation with Endo H confirmed the efficient complex glycosylation and revealed the four extracellular N-linked glycosylation sites.
Effects of β subunits on the current amplitude of hNav1.8
To first confirm that the β subunits exerted the expected effects upon expression in Xenopus oocytes, their modulation of Nav1.2 was first investigated. As shown previously [Citation44,Citation45], the β1 and β3 subunits shifted voltage-dependence of inactivation in the hyperpolarising direction and accelerated current decay kinetics of rNav1.2, whereas the β2 subunit was without effect (Supplementary Fig. 1).
After establishing that the β subunits had the expected effects on rNav1.2, we investigated their effect on hNav1.8. Expression of hNav1.8 (2 ng/oocyte) alone and with the β1, β2 or β3 subunits (5 ng cRNA/oocyte), corresponding to an α:β ratio of 1:2.5. The β1 and β2 subunits did not affect the Na+ current amplitude of hNav1.8, however, the β3 subunit caused a pronounced decrease in maximal Na+ current amplitude (Imax) to 22 ± 4% of control (n = 40; p ≤ 0.001) (oocytes expressing only hNav1.8; n = 56; average Na+ current amplitude 0.79 ± 0.38 μA) ()).
Effects of β subunits on current kinetics/channel properties
We then determined the effects of the β subunits on the biophysical properties of hNav1.8, again at an α:β ratio of 1:2.5 (). The β1 subunit caused a hyperpolarizing shift of 6.9 mV in the activation curve of hNav1.8 ()); the value for V0.5 (activation) was significantly different between these two groups (p ≤ 0.001; ). The β1 subunit also significantly altered voltage-dependence of inactivation of hNav1.8 by shifting V0.5 10.68 mV in the hyperpolarizing direction (p ≤ 0.001; ) and ). Furthermore, the β1 subunit accelerated the inactivation kinetics of hNav1.8 (single exponential fits, p ≤ 0.1; ) and ).
We next investigated whether the β subunits modulated recovery from inactivation (repriming). Recovery from inactivation of hNav1.8 occurs in two phases, as has been reported previously [Citation36]. The β3 subunit strongly decelerated recovery from inactivation whereas the other β subunits were without effect ()). In the presence of the β3 subunit, both τ1 (τfast) and τ2 (τslow) were significantly slower than for hNav1.8 expressed alone (p ≤ 0.01 and p ≤ 0.0001 for τ1 and τ2, respectively, ). The percentage of current recovering with fast kinetics was also significantly lower in the presence of the β3 subunit than in the other three groups (p ≤ 0.0001; ).
To determine whether the effect of the β3 subunit on Imax of hNav1.8 was dependent on the α:β3 ratio, different amounts of β3 cRNA (0.5, 1 or 5 ng) were injected into each oocyte in the presence of the same amount of hNav1.8 cRNA (2.5 ng), corresponding to α:β3 ratios of 1:0.25, 1:0.4 or 1:25, respectively. We found that the effect of the β3 subunit on hNav1.8 maximal current amplitude was dependent on the α:β3 ratio ()). Similarly to the down-regulation of current amplitude by the β3 subunit, the effects on repriming were dependent on the α:β3 ratio ()).
Functional comparison of rat and human β3 subunits
The β subunits used in this study were of rat origin. Rat and human β1 subunits share 93% homology in their amino acid sequence, rat/human β2 subunits share 96% homology and rat/human β3 subunits are 98% homologous. The high degree of homology suggests that the modulation of VGSC α-subunits does not differ between rat and human β subunits. The most striking modulation of hNav1.8 was mediated by the β3 subunit. To verify that the human and rat β3 subunits mediated similar effects on hNav1.8, the four amino acids of rat β3 that differ were mutated to the corresponding residues in the human β3 subunit (L8F, R20S, F174L and V210A). When co-expressed with hNav1.8, rβ3[L8F, R20S, F174L, V210A] caused almost identical effects to wild-type rat β3 on current amplitude and recovery from inactivation of hNav1.8 (), suggesting that rat and human β3 modulate hNav1.8 in a similar manner.
Effects of auxiliary β1/β3 subunits chimeras on hNav1.8
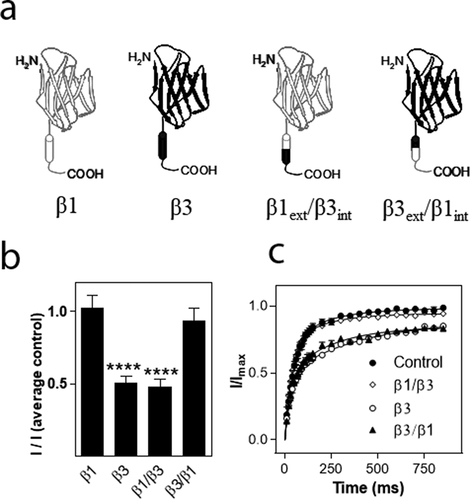
To investigate which part of the β3 subunit mediated the effects on hNav1.8 current amplitude and recovery from inactivation, respectively, we created chimeras of the β1 and β3 subunit in which the extracellular domain of β1 was combined with the intracellular domain of β3 (β1ext/β3int) or vice versa (β3ext/β1int). The hNav1.8 α subunit was then co-expressed with the chimeric β subunits in a 1:1 ratio. For this series of experiments, the current mediated by hNav1.8 expressed alone exhibited an Imax of 0.82 ± 0.04 µA (n = 172). As for the previous series of experiments, the β1 subunit had no significant effect on the Imax of Nav1.8, whereas co-expression with the β3 subunit caused a significant decrease (p < 0.001) of the Nav1.8 current amplitude (n > 9). The β1ext/β3int chimera caused a similar reduction of hNav1.8 Imax to that observed for the β3 subunit, whereas the β3ext/β1int chimera subunit with hNav1.8, did not significantly reduce Imax when compared to the hNav1.8 control ()). For these series of experiments, 2.5 ng of each cRNA was injected into each oocyte. Thus, the α:β3 ratio was 1:1 rather than 2:5, explaining why the amplitude was not reduced to the same extent as what is shown in ). Similarly, when hNav1.8 was co-expressed with both β1 and β3, the ratio was 1:0.5:0.5, effectively diluting the reducing effect of the β3 subunit in comparison to the result shown in ).
We then assessed the effects of the chimeric β subunits on the recovery from inactivation of hNav1.8. The β3 subunit as well as the β1ext/β3int chimera significantly decelerated recovery from inactivation (p < 0.01) whereas the β3ext/β1int chimera had no effect on recovery from inactivation ()). Interestingly, whereas the β3 subunit decelerated τ of both the fast and the slow phase, the β1ext/β3int chimera increased τ2 by approximately 30 ms (from 54 ± 5 ms to 88 ± 16 ms; n ≥ 20) but did not significantly alter τ1. Co-expression of hNav1.8 with the β3 subunit or the β1ext/β3int chimera only allowed 90% recovery of the maximum current obtained within a second, whereas full recovery was seen within 5 s of all the combinations used.
Discussion
This study reports on the modulation of human Nav1.8 expressed in Xenopus oocytes by auxiliary β subunits. Nav1.8 is implicated in pain states and remains a promising target for drug discovery, for which the Xenopus oocyte expression system is a valuable screening platform [Citation38]. To better mimic the natural environment in DRG neurons, Nav1.8 can be co-expressed with β subunits in Xenopus oocytes. Therefore, it is important to determine how β subunits modulate hNav1.8 in this system.
When expressed in Xenopus oocytes, hNav1.8 mediated a Na+ current with the TTX-resistance and slow kinetics characteristic of Nav1.8 in native DRG neurons [Citation12,Citation13] as has been shown previously [Citation33]. The gating properties of hNav1.8 in the absence of β subunits was similar to human Nav1.8 expressed in Xenopus oocytes [Citation33], with minor differences from that previously reported for rat Nav1.8 [Citation5,Citation30]. Repriming kinetics were similar to that previously described for human Nav1.8 in mammalian cells [Citation36]. The β1 subunit accelerated current decay kinetics for hNav1.8, as has been described previously for rNav1.8 [Citation30,Citation46]. β1 also caused hyperpolarizing shifts of both voltage-dependence of activation and inactivation of hNav1.8. Similar shifts in voltage-dependence of activation and inactivation mediated by the β1 subunit have been reported for rNav1.8 expressed in Xenopus oocytes [Citation5,Citation30,Citation46] and mammalian cells [Citation37]. In the present study, we did not find any effects of the β3 subunit on the voltage-dependence of activation/inactivation. This is consistent with data from rNav1.8 in mammalian cells [Citation37], however, β3 expressed with rNav1.8 in Xenopus oocytes shifted both curves in the hyperpolarizing direction [Citation47] or shifted the inactivation curve in the depolarizing direction [Citation46].
The most pronounced effects of any of the auxiliary β subunits on hNav1.8 were the modulation of recovery from inactivation and maximal Na+ current amplitude by the β3 subunit. β3 markedly decelerated the repriming kinetics of hNav1.8 and reduced maximal Na+ current amplitude to ~25% of control levels (α:β3 ratio 1:2.5). This modulation of repriming resembled the effect of lidocaine and related compounds on VGSCs, including Nav1.8 [Citation48]. Delayed recovery from inactivation mediated by the β subunits has not been reported previously. Instead, other studies have shown that the β3 subunit can accelerate repriming of VGSCs, Nav1.5 [Citation49] and Nav1.3 [Citation5,Citation50]. Whilst we did not find that the β1 subunit modulated recovery from inactivation, a previous study has shown that β1 accelerated the first phase but decelerated the second phase of rat Nav1.8 repriming [Citation30].
Unlike the β1 and β2 subunits, β3 has been shown to significantly reduce the current density of Nav1.8 [Citation37]. This correlates well with our observation showing that β3 down-regulates the maximal current amplitude. However, two other studies report an up-regulation of rNav1.8 current amplitude associated with β3 co-expression in Xenopus oocytes [Citation5,Citation46]. Differences in cRNA concentration and/or incubation time post-injection between studies may contribute to these differences, along with differences in human and rat Nav1.8, which are only 82% homologous. Furthermore, whilst we did not find that the β1 subunit affected the current amplitude of hNav1.8, other studies on rNav1.8 in Xenopus oocytes [Citation46] and mammalian cells [Citation37] have reported that β1 can increase current amplitude/current density. Again, discrepancies can be due to differences in the precise experimental design or expression of endogenous factors between expression systems. Therefore, it is a priority to evaluate the effect of the β subunits on Nav1.8 in native sensory neurons and a study of the expression of human and rat Nav1.8 in DRG neurons revealed subtle differences in the biophysical properties of the channels [Citation51].
VGSC β subunits can function as cell adhesion molecules (CAMs), playing important roles in cell-cell adhesion. β subunits also modulate cell surface levels of VGSCs, most likely via anchoring the VGSC to the cytoskeleton [Citation52,Citation53]. Thus, the modulation of hNav1.8 current amplitude by the β3 subunit may involve regulation by several mechanisms including modulation of channel opening probability, stabilization of the channel in the plasma membrane, cross-linking with other α or β subunits, alterations in trafficking or, less likely, signaling events leading to altered mRNA levels. Single-channel patch-clamp recording experiments in mammalian cells could be used to determine whether the β3 subunit directly alters the opening probability of hNav1.8.
The VGSC is believed to exist in vivo as a heterodimer or heterotrimer consisting of one α subunit and one or two β subunits. Traditionally, it was considered that one α subunit can interact with one non-covalently linked (β1 or β3) and one disulfide-linked β subunit (β2 or β4) [Citation2,Citation54]. Recent studies suggest that interactions between VGSC α and β subunits are far more complex. One recent study, which investigated the structure of the immunoglobulin (Ig) domain of the β3 subunit using crystallography and single-molecule resolution imaging, reported that this domain assembles as a trimer. This study also reported that the β3 subunit can bind to multiple sites on the Nav1.5 α-subunit and induce the formation of α subunit oligomers, possibly resulting in cross-linking of multiple α and β subunits [Citation41]. In the current study, we observed that the key effects of β3 appeared to be dependent on the α:β3 ratio (or the amount of β3 cRNA injected). It is possible that the number of β3 subunits in the membrane was not enough to saturate all Nav1.8 α subunits when the lower β3 cRNA concentrations were injected, however, more complex interactions such as β3 subunit crosslinking could also be involved. To date, we do not know whether such interactions modulate cell surface levels or biophysical properties of VGSCs.
It was previously thought that VGSC α and β subunits primarily interacted via their extracellular domains, however, more recent findings have demonstrated that their intracellular domains also interact. A recent study reported on the cryo-EM structure of the electric eel Nav1.4 α-subunit (EeNav1.4) in complex with the β1 subunit. This study showed that the extracellular Ig domain of β1 docks with extracellular loop 5 (from domain I) and loop 6 (from domain IV) of the α-subunit, whereas the β1 transmembrane helix interacts with the third voltage-sensing domain (VSDIII) of the α subunit [Citation40]. Our data obtained using chimeras of the β1 and β3 subunits show that the extracellular domain of the β3 subunit mediated the effects on recovery from inactivation, whereas the down-regulation in current amplitude was modulated by the intracellular domain of the β3 subunit. Nav1.8 can interact with several intracellular proteins including cytoskeletal proteins, channel-associated proteins, motor proteins and enzymes which may regulate Nav1.8 membrane density [Citation55]. In particular, annexin light chain (p11) is a strong regulator of trafficking and cell surface levels of Nav1.8 [Citation56]. Ubiquitination and subsequent proteasomal degradation have also been shown to potently regulate cell surface levels of Nav1.8 [Citation57]. It is thus possible that the β3 subunit interferes with another intracellular regulatory protein, indirectly modulating Nav1.8. In regard to recovery from inactivation, it is possible that interactions between the extracellular domains of Nav1.8 and the β3 subunit indirectly modulate sites in Nav1.8 involved in repriming, such as the transmembrane S6 segment [Citation49] or the S3-S4 linker of domain IV [Citation58].
All four known VGSC β subunits are expressed in sensory neurons [Citation14,Citation29,Citation59]. The β3 subunit is the main β subunit expressed in nociceptive neurons and is therefore most likely to modulate VGSC behavior in these neurons [Citation5,Citation28,Citation60]. Both the β1 and β3 subunits appear to play a role in pain, since they are up-regulated in rat and human DRG in neuropathic pain states [Citation5,Citation61]. The main role of the β1 and β3 subunits in alteration of DRG Na+ current profiles in neuropathic pain appears to be due to interactions with the VGSC subunit induced in neuropathic pain states. Heterologous expression experiments in Xenopus oocytes and mammalian cells have demonstrated that both the β1 and β3 subunits can further accelerate the already rapid repriming kinetics of Nav1.3, possibly promoting repetitive firing. In addition, β1 and β3 subunits lower the activation threshold of Nav1.3, thereby further contributing to increased excitability [Citation5,Citation50]. β3 has also been shown to co-localize with Nav1.7 in small dorsal root ganglion neurons and when co-expressed in mammalian cells, β3 modulated the gating properties of Nav1.7 with hyperpolarizing and depolarizing shifts in activation and inactivation, respectively. β3 also accelerated recovery from inactivation; together, these alterations may increase neuronal excitability [Citation60].
The reduction of Nav1.8 current amplitude observed in the present study, consistent with a previous study in mammalian cells [Citation37], would theoretically decrease the activity of Nav1.8 when translated into an in vivo situation. Furthermore, a decelerated recovery from inactivation could decrease the opening probability of the channel. The down-regulation in TTX-R Na+ current and the up-regulation of the β3 subunit in the DRG in neuropathic pain states are thought to be two independent phenomena that are caused by alterations in growth factor levels [Citation62–65]. However, the data presented in the current and previous study [Citation37] present a mechanism to explain how these two phenomena may be interrelated. If this is the case, the increased levels of the β3 subunit may contribute to the suppression of TTX-R Na+ current observed in neuropathic pain states.
Figure 1. Expression of human Nav1.8 and rat Nav1.2 in Xenopus oocytes. (a) When expressed in Xenopus oocytes, hNav1.8 mediates an inward Na+ current with slow activation and inactivation kinetics that is unaffected by 1 μM tetrodotoxin (TTX). (b) In contrast, rat Nav1.2 mediates a Na+ current exhibiting fast activation and inactivation kinetics that is completely abolished by the application of 1 μM TTX. Oocytes were held at – 70 mV and depolarized to voltages between – 50 and +40 mV in 10 mV increments. External solutions containing TTX (1 µM) were applied through the perfusion system
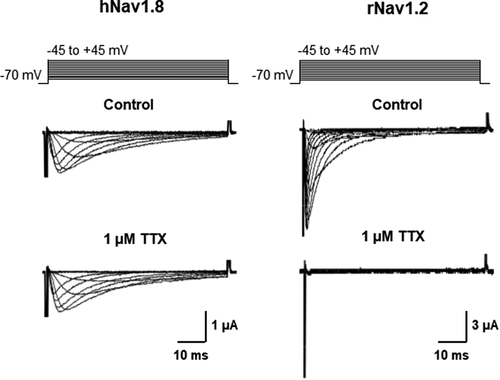
Figure 2. Biochemical analysis of the synthesis and plasma membrane transport of the sodium channel β3 subunit in Xenopus laevis oocytes. Oocytes injected with cRNA encoding the His-tagged β3 subunit or non-injected controls (c) were metabolically labeled with [35S]-methionine (left panel) or surface-iodinated with [125I]-sulfo-SHPP (right panel). His-tagged protein was purified via Ni2+-NTA-agarose, treated with endoglycosidases (concentrations given in IUB milliunits/ml sample) as indicated, and separated on a 10% SDS-PAGE gel. Black and white triangles indicate complex glycosylated and completely deglycosylated protein, respectively. Numbers 1–4 indicate the Endo H-sensitive core-glycosylated and partly deglycosylated forms of the protein
![Figure 2. Biochemical analysis of the synthesis and plasma membrane transport of the sodium channel β3 subunit in Xenopus laevis oocytes. Oocytes injected with cRNA encoding the His-tagged β3 subunit or non-injected controls (c) were metabolically labeled with [35S]-methionine (left panel) or surface-iodinated with [125I]-sulfo-SHPP (right panel). His-tagged protein was purified via Ni2+-NTA-agarose, treated with endoglycosidases (concentrations given in IUB milliunits/ml sample) as indicated, and separated on a 10% SDS-PAGE gel. Black and white triangles indicate complex glycosylated and completely deglycosylated protein, respectively. Numbers 1–4 indicate the Endo H-sensitive core-glycosylated and partly deglycosylated forms of the protein](/cms/asset/6a45c0ca-d05b-4d5a-a054-b94eeac92819/kchl_a_1860399_f0002_b.gif)
Figure 3. Modulation of hNav1.8 by auxiliary β subunits. (a) Effects of β subunits on Na+ current amplitude. I represents maximal Na+ current amplitude of oocytes expressing hNav1.8 (2 ng cRNA/oocyte) alone or in combination with β1, β2 or β3 (5 ng cRNA/oocyte). I(average control) represents the average maximal Na+ current amplitude of oocytes expressing only hNav1.8. Maximal Na+ current amplitude was determined by step depolarizations to voltages between – 50 and +50 mV (5 mV increments) from a holding potential of – 70 mV. The voltages at which maximal Na+ current amplitude was obtained was +5 mV for hNav1.8 + β1 and +10 mV for the other combinations (including Nav1.8 in the absence of β subunits). Curves show the Na+ conductance (G) obtained at different voltages relative to the maximal conductance (Gmax). Conductance curves were fitted with single exponential functions for the hNav1.8 α subunit alone and in the presence of the various β subunits. (c) Voltage-dependence of inactivation. I represents the Na+ current elicited by a depolarizing pulse to the voltage generating maximal Na+ current amplitude immediately after long (1 s) pre-pulses to different voltages. I–120 represents the Na+ current amplitude elicited by an identical depolarizing pulse generated after a long pre-pulse to – 120 mV, where inactivation is minimal. I/I−120 represents the fraction of maximal Na+ current available after steady-state inactivation at each voltage. (d) Inactivation kinetics. Superimposed traces normalized to the same value are shown for Na+ currents mediated by hNav1.8 in the absence and presence of the β1, β2 and β3 subunit (5 ng cRNA/oocyte). Oocytes were held at – 70 mV and depolarized to the voltage that elicited maximal Na+ current amplitude. (e) Recovery from inactivation. The fraction of Na+ current recovering from steady-state inactivation after different periods of time (2.5 ms – 1 s) was determined for hNav1.8 (2.5 ng cRNA/oocyte) expressed alone or together with the β1, β2 or β3 subunit (5 ng cRNA/oocyte). Na+ current was first inactivated by a 1 s pulse to 0 mV. After a variable recovery period ranging from 2.5 ms – 1 s, a depolarizing pulse to elicit maximal Na+ current amplitude was applied. The Na+ current amplitude after different recovery times (I) was compared to the Na+ current amplitude elicited by an identical control pulse that was not preceded by inactivation (Imax). The recovered fraction of Na+ current (I/Imax) was plotted against recovery time and fitted with double exponential functions
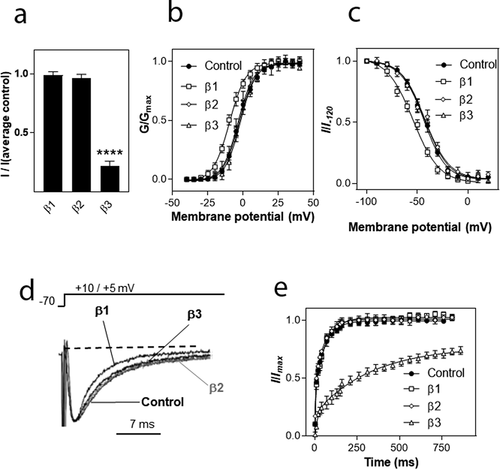
Figure 4. Effects of the α:β3 ratio on β3-mediated modulation of the hNav1.8 current amplitude and recovery from inactivation. (a) Effects of the α:β3 ratio on current amplitude. Maximal Na+ current amplitude was recorded from oocytes injected with cRNA for hNav1.8 (2.5 ng/oocyte) alone or together with 0.5, 1, or 5 ng of cRNA encoding the β3-subunit. I represents maximal Na+ current amplitude in the various groups while I(average control) represents the average maximal Na+ current amplitude of control (oocytes expressing only hNav1.8). N = 30–41 oocytes/group. (b) Effects of the α:β3 ratio on the repriming kinetics of hNav1.8. Recovery from inactivation was determined as described for hNav1.8 alone. The Na+ current amplitude after different recovery times (i) was compared to the Na+ current amplitude generated by an identical control pulse (Imax). The repriming curves were fitted with double exponential functions (N ≥ 10 oocytes/group)
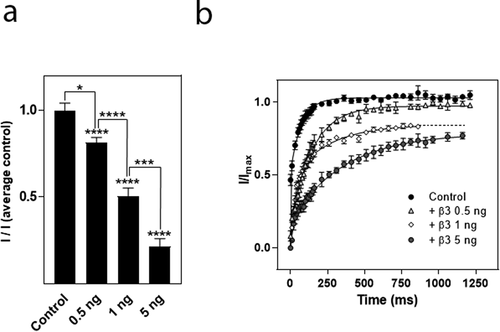
Figure 5. Comparison of modulation of hNav1.8 by the rat and human β3 subunits. (a). Effects on Na+ current amplitude. Maximal Na+ current amplitude was determined for oocytes expressing hNav1.8 alone or in combination with the rat or human β3-subunit. I represents the maximal Na+ current amplitude of oocytes expressing hNav1.8 alone or in combination with the β3 subunit. I(average control) represents the average maximal Na+ current amplitude of oocytes expressing only hNav1.8. ****significantly different from control, p ≤ 0.0001. (b) Comparison of the modulation of recovery from inactivation of hNav1.8 by the rat and human β3 subunits (N = 15–23 oocytes/group)
Figure 6. Effects of β subunits chimeras on maximal current amplitude and recovery from inactivation of hNav1.8. (a). Schematic showing the structure of the wild-type β1 and β3 subunits and the constructed chimeras (β1: white, β3: black). (b) Effects on Na+ current amplitude. Maximal Na+ current amplitude was determined for oocytes expressing hNav1.8 alone or in combination with the rat β3 chimera subunits. I represents maximal Na+ current amplitude of oocytes expressing hNav1.8 alone or in combination with the β3 chimera subunits
Supplemental Material
Download MS Word (14.1 KB)Supplemental Material
Download MS Word (77.2 KB)Acknowledgments
We thank Dr Jenny Ekberg (Griffith University) for her contribution to this study. We also thank Jenny Kastberg for assistance with plasmid cloning and members of the Adams and Lewis laboratories for technical assistance regarding this project.
Disclosure statement
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Hartshorne RP, Messner DJ, Coppersmith JC, et al. The saxitoxin receptor of the sodium channel from rat brain. Evidence for two nonidentical β subunits. J Biol Chem. 1982;257(23):13888–13891.
- Hartshorne RP, Catterall WA. The sodium channel from rat brain. Purification and subunit composition. J Biol Chem. 1984;259(3):1667–1675.
- Morgan K, Stevens EB, Shah B, et al. β3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci U S A. 2000;97(5):2308–2313.
- Yu FH, Westenbroek RE, Silos-Santiago I, et al. Sodium channel β4, a new disulfide-linked auxiliary subunit with similarity to β2. J Neurosci. 2003;23(20):7577–7585.
- Shah BS, Stevens EB, Gonzalez MI, et al. β3, a novel auxiliary subunit for the voltage-gated sodium channel, is expressed preferentially in sensory neurons and is upregulated in the chronic constriction injury model of neuropathic pain. Eur J Neurosci. 2000;12(11):3985–3990.
- Isom LL, De Jongh KS, Patton DE, et al. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256(5058):839–842.
- Isom LL, Ragsdale DS, De Jongh KS, et al. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83(3):433–442.
- Kraner SD, Tanaka JC, Barchi RL. Purification and functional reconstitution of the voltage-sensitive sodium channel from rabbit T-tubular membranes. J Biol Chem. 1985;260(10):6341–6347.
- Black JA, Dib-Hajj S, McNabola K, et al. Spinal sensory neurons express multiple sodium channel α-subunit mRNAs. Brain Res Mol Brain Res. 1996;43(1–2)):117–131.
- Felts PA, Yokoyama S, Dib-Hajj S, et al. Sodium channel α-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res Mol Brain Res. 1997;45(1):71–82.
- Dib-Hajj SD, Tyrrell L, Black JA, et al. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci U S A. 1998;95(15):8963–8968.
- Djouhri L, Fang X, Okuse K, et al. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol. 2003;550(Pt 3):739–752.
- Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379(6562):257–262.
- Sutkowski EM, Catterall WA. β1 subunits of sodium channels. Studies with subunit-specific antibodies. J Biol Chem. 1990;265(21):12393–12399.
- Akopian AN, Souslova V, England S, et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2(6):541–548.
- Wang J, Ou S-W, Wang Y-J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels. 2017;11(6):534–554.
- Faber CG, Lauria G, Merkies IS, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci USA. 2012;109(47):19444–19449.
- Zimmermann K, Leffler A, Babes A, et al. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447(7146):855–858.
- Dib-Hajj SD, Binshtok AM, Cummins TR, et al. Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment. Brain Res Rev. 2009;60(1):65–83.
- Porreca F, Lai J, Bian D, et al. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci U S A. 1999;96(14):7640–7644.
- Novakovic SD, Tzoumaka E, McGivern JG, et al. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci. 1998;18(6):2174–2187.
- Okuse K, Chaplan SR, McMahon SB, et al. Regulation of expression of the sensory neuron-specific sodium channel SNS in inflammatory and neuropathic pain. Mol Cell Neurosci. 1997;10(3/4):196–207.
- Dib-Hajj SD, Fjell J, Cummins TR, et al. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain. 1999;83(3):591–600.
- Dib-Hajj S, Black JA, Felts P, et al. Down-regulation of transcripts for Na channel α-SNS in spinal sensory neurons following axotomy. Proc Natl Acad Sci USA. 1996;93(25):14950–14954.
- Coward K, Jowett A, Plumpton C, et al. Sodium channel β1 and β2 subunits parallel SNS/PN3 α-subunit changes in injured human sensory neurons. Neuroreport. 2001;12(3):483–488.
- Meadows LS, Chen YH, Powell AJ, et al. Functional modulation of human brain Nav1.3 sodium channels, expressed in mammalian cells, by auxiliary β1, β2 and β3 subunits. Neuroscience. 2002;114(3):745–753.
- Blackburn-Munro G, Fleetwood-Walker SM. The sodium channel auxiliary subunits β1 and β2 are differentially expressed in the spinal cord of neuropathic rats. Neuroscience. 1999;90(1):153–164.
- Casula MA, Facer P, Powell AJ, et al. Expression of the sodium channel β3 subunit in injured human sensory neurons. Neuroreport. 2004;15(10):1629–1632.
- Takahashi N, Kikuchi S, Dai Y, et al. Expression of auxiliary β subunits of sodium channels in primary afferent neurons and the effect of nerve injury. Neuroscience. 2003;121(2):441–450.
- Vijayaragavan K, O’Leary ME, Chahine M. Gating properties of Nav1.7 and Nav1.8 peripheral nerve sodium channels. J Neurosci. 2001;21(20):7909–7918.
- Fitzgerald EM, Okuse K, Wood JN, et al. cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS. J Physiol. 1999;516(Pt 2):433–446.
- John VH, Main MJ, Powell AJ, et al. Heterologous expression and functional analysis of rat Nav1.8 (SNS) voltage-gated sodium channels in the dorsal root ganglion neuroblastoma cell line ND7-23. Neuropharmacology. 2004;46(3):425–438.
- Zhang Z-N, Li Q, Liu C, et al. The voltage-gated Na+ channel Nav1.8 contains an ER-retention/retrieval signal antagonized by the β3 subunit. J Cell Sci. 2008;121:3243–3252.
- Knapp O, Nevin ST, Yasuda T, et al. Biophysical properties of Nav1.8/Nav1.2 chimeras and inhibition by µO-conotoxin MrVIB. Br J Pharmacol. 2012;166(7):2148–2160.
- Deuis JR, Dekan Z, Inserra MC, et al. Development of a µO-conotoxin analogue with improved lipid membrane interactions and potency for the analgesic sodium channel Nav1.8. J Biol Chem. 2016;291(22):11829–11842.
- Browne LE, Clare JJ, Wray D. Functional and pharmacological properties of human and rat Nav1.8 channels. Neuropharmacology. 2009;56(5):905–914.
- Zhao J, O’Leary ME, Chahine M. Regulation of Nav1.6 and Nav1.8 peripheral nerve Na+ channels by auxiliary β-subunits. J Neurophysiol. 2011;106(2):608–619.
- Kvist T, Hansen KB, Brauner-Osborne H. The use of Xenopus oocytes in drug screening. Expert Opin Drug Discov. 2011;6(2):141–153.
- Messner DJ, Catterall WA. The sodium channel from rat brain. Separation and characterization of subunits. J Biol Chem. 1985;260(19):10597–10604.
- Yan Z, Zhou Q, Wang L, et al. Structure of the Nav1.4-β1 complex from electric eel. Cell. 2017;170(3):470–482e11.
- Namadurai S, Balasuriya D, Rajappa R, et al. Crystal structure and molecular imaging of the Nav channel β3 subunit indicates a trimeric assembly. J Biol Chem. 2014;289(15):10797–10811.
- Gloor S, Pongs O, Schmalzing G. A vector for the synthesis of cRNAs encoding Myc epitope-tagged proteins in Xenopus laevis oocytes. Gene. 1995;160(2):213–217.
- Nicke A, Bäumert HG, Rettinger J, et al. P2X1 and P2X3 receptors form stable trimers: a novel structural motif of ligand-gated ion channels. Embo J. 1998;17(11):3016–3028.
- Smith RD, Goldin AL. Functional analysis of the rat I sodium channel in Xenopus oocytes. J Neurosci. 1998;18(3):811–820.
- Stevens EB, Cox PJ, Shah BS, et al. Tissue distribution and functional expression of the human voltage-gated sodium channel β3 subunit. Pflugers Arch. 2001;441(4):481–488.
- Vijayaragavan K, Powell AJ, Kinghorn IJ, et al. Role of auxiliary β1-, β2-, and β3-subunits and their interaction with Nav1.8 voltage-gated sodium channel. Biochem Biophys Res Commun. 2004;319(2):531–540.
- Wilson MJ, Zhang MM, Azam L, et al. Navβ subunits modulate the inhibition of Nav1.8 by the analgesic gating modifier µO-conotoxin MrVIB. J Pharmacol Exp Ther. 2011;338(2):687–693.
- Browne LE, Blaney FE, Yusaf SP, et al. Structural determinants of drugs acting on the Nav1.8 channel. J Biol Chem. 2009;284(16):10523–10536.
- Zhu W, Voelker TL, Varga Z, et al. Mechanisms of noncovalent β subunit regulation of NaV channel gating. J Gen Physiol. 2017;149(8):813–831.
- Cummins TR, Aglieco F, Renganathan M, et al. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci. 2001;21(16):5952–5961.
- Han C, Estacion M, Huang J, et al. Human Nav1.8: enhanced persistent and ramp currents contribute to distinct firing properties of human DRG neurons. J Neurophysiol. 2015;113(9):3172–3185.
- Isom LL. Sodium channel β subunits: anything but auxiliary. Neuroscientist. 2001;7(1):42–54.
- Isom LL. The role of sodium channels in cell adhesion. Front Biosci. 2002;7:12–23.
- Barchi RL, Casadei JM, Gordon RD, et al. Voltage-sensitive sodium channels: an evolving molecular view. Soc Gen Physiol Ser. 1987;41:125–148.
- Malik-Hall M, Poon WY, Baker MD, et al. Sensory neuron proteins interact with the intracellular domains of sodium channel NaV1.8. Brain Res Mol Brain Res. 2003;110(2):298–304.
- Okuse K, Malik-Hall M, Baker MD, et al. Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature. 2002;417(6889):653–656.
- Fotia AB, Ekberg J, Adams DJ, et al. Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J Biol Chem. 2004;279(28):28930–28935.
- Dib-Hajj SD, Ishikawa K, Cummins TR, et al. Insertion of a SNS-specific tetrapeptide in S3-S4 linker of D4 accelerates recovery from inactivation of skeletal muscle voltage-gated Na channel µ1 in HEK293 cells. FEBS Lett. 1997;416(1):11–14.
- Oh Y, Sashihara S, Black JA, et al. Na+ channel β1 subunit mRNA: differential expression in rat spinal sensory neurons. Brain Res Mol Brain Res. 1995;30(2):357–361.
- Ho C, Zhao J, Malinowski S, et al. Differential expression of sodium channel β subunits in dorsal root ganglion sensory neurons. J Biol Chem. 2012;287(18):15044–15053.
- Blackburn-Munro G, Fleetwood-Walker SM. The sodium channel β3 subunit in injured human sensory neurons. Neuroreport. 2004;15(1):153–164.
- Fjell J, Cummins TR, Dib-Hajj SD, et al. Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Brain Res Mol Brain Res. 1999;67(2):267–282.
- Leffler A, Cummins TR, Dib-Hajj SD, et al. GDNF and NGF reverse changes in repriming of TTX-sensitive Na+ currents following axotomy of dorsal root ganglion neurons. J Neurophysiol. 2002;88(2):650–658.
- Cummins TR, Black JA, Dib-Hajj SD, et al. Glial-derived neurotrophic factor upregulates expression of functional SNS and NaN sodium channels and their currents in axotomized dorsal root ganglion neurons. J Neurosci. 2000;20(23):8754–8761.
- Dib-Hajj SD, Black JA, Cummins TR, et al. Rescue of α-SNS sodium channel expression in small dorsal root ganglion neurons after axotomy by nerve growth factor in vivo. J Neurophysiol. 1998;79(5):2668–2676.