
ABSTRACT
Leydig cells are the principal steroidogenic cells of the testis. Leydig cells also secrete a number of growth factors including vascular endothelial growth factor (VEGF) which has been shown to regulate both testicular steroidogenesis and spermatogenesis. The thyroid hormone, T3, is known to stimulate steroidogenesis in Leydig cells. T3 has also been shown to stimulate VEGF production in a variety of cell lines. However, studies regarding the effect of T3 on VEGF synthesis and secretion by the Leydig cells were lacking. Therefore, we investigated the effect of T3 on VEGF synthesis and secretion in a mouse Leydig tumour cell line, MLTC-1. The effect of T3 was compared with that of LH/cAMP and hypoxia, two known stimulators of Leydig cell functions. The cells were treated with T3, 8-Br-cAMP (a cAMP analogue), or CoCl2 (a hypoxia mimetic) and VEGF secreted in the cell supernatant was measured using ELISA. The mRNA levels of VEGF were measured by quantitative RT-PCR. In the MLTC-1 cells, T3, 8-Br-cAMP, and CoCl2 stimulated VEGF mRNA levels and the protein secretion. T3 also increased steroid secretion as well as HIF-1α protein levels, two well-established upstream regulators of VEGF. Inhibitors of steroidogenesis as well as HIF-1α resulted in inhibition of T3-stimulated VEGF secretion by the MLTC-1 cells. This suggested a mediatory role of steroids and HIF-1α protein in T3-stimulated VEGF secretion by MLTC-1 cells. The mediation by steroids and HIF-1α were independent of each other.
Abbreviations: 8-Br-cAMP: 8-bromo – 3ʹ, 5ʹ cyclic adenosine monophosphate; CoCl2: cobalt chloride; HIF-1α: hypoxia inducible factor −1α; LH: luteinizing hormone; T3: 3, 5, 3ʹ-L-triiodothyronine; VEGF: vascular endothelial growth factor
KEYWORDS:
Introduction
Leydig cells are the principal steroidogenic cells of the testis. Leydig cell functions are primarily regulated by luteinizing hormone (LH) and oxygen tension. LH and its second messenger, 3ʹ, 5ʹ cyclic adenosine monophosphate (cAMP) are known to stimulate steroid production and secretion from Leydig cells and their derived lines. In contrast, low oxygen tension or hypoxia have been shown to inhibit steroid production and secretion from Leydig cell derived MA-10 cells (Kumar et al. Citation2014).
In addition to steroids, Leydig cells and their derived lines secrete a number of growth factors including vascular endothelial growth factor (VEGF) (Anand et al. Citation2003; Schwarzenbach et al. Citation2004; Kumar et al. Citation2012). VEGF, in turn, stimulates Leydig cell proliferation and testosterone secretion (Hwang et al. Citation2007). VEGF also regulates spermatogenesis by stimulating proliferation and self-renewal of spermatogonial cells as well as by promoting their differentiation (Caires et al. Citation2012; Sargent et al. Citation2016).
The prime regulators of Leydig cell function, LH and hypoxia, also regulate VEGF synthesis and secretion from Leydig cells. In primary Leydig cells and MA-10 cells, both LH and hypoxia were shown to stimulate VEGF synthesis and secretion (Anand et al. Citation2003; Schwarzenbach et al. Citation2004; Kumar et al. Citation2012). In MA-10 cells, CoCl2-induced hypoxia was shown to stimulate VEGF secretion through the mediation of the transcription factor, hypoxia inducible factor-1α (HIF-1α) (Kumar et al. Citation2012). Steroids secreted by the Leydig cells also act in an autocrine manner to stimulate VEGF production and secretion. In MA-10 cells, progesterone was shown to stimulate VEGF expression and secretion (Schwarzenbach et al. Citation2004).
Another important regulator of Leydig cell function are the thyroid hormones. Thyroid hormone receptors have been identified on the Leydig cells (Jana and Bhattacharya Citation1994). Thyroid hormone, 3, 5, 3ʹ-L-triiodothyronine (T3) has been shown to increase basal and cAMP-stimulated steroid production in primary Leydig cells and their derived lines (Manna et al. Citation1999, Citation2001; Maran et al. Citation2000). T3 also stimulates VEGF mRNA and protein levels in a variety of other cell lines including rat brain-derived endothelial cells and human hepatoma cells (Zhang et al. Citation2010; Chen et al. Citation2014). Hyperthyroid patients also have higher serum VEGF levels (Iitaka et al. Citation1998). However, no studies have been conducted to see the direct modulation of VEGF expression and secretion by T3 in Leydig cells. Therefore, in the present study we investigated the effect of T3 on the synthesis of VEGF by the mouse Leydig tumour cell line, MLTC-1 and also studied the mediatory pathway(s).
Results
Time course and dose response of T3 on VEGF secretion
T3 stimulated VEGF secretion in a time dependent manner as shown in . A significant difference in VEGF secretion with T3 was observed from 6 hours onwards. However, by 9 hours MLTC-1 cells began to proliferate (data not shown). Therefore, all further treatments in MLTC-1 cells were done for 6 hours, before the cells began to divide. No difference was found in the morphology of the cells with and without treatment with T3 as observed under a microscope.
Figure 1. Time course and dose response of VEGF secretion in response to T3 by MLTC-1 cells. (A) Time course of VEGF secretion in response to 2.5 ng/ml of T3. Lines represent mean ± SD of three experiments; each experiment was done in triplicate (n = 9). ** p < 0.01; *** p < 0.001 between untreated and T3 treated cells at respective time points. (B) Dose response of T3 on VEGF secretion. MLTC-1 cells were incubated for 6 hours with different doses of T3. Bars represent mean ± SD of three experiments; each experiment was done in triplicate (n = 9). * p < 0.05; *** p < 0.001 between untreated and different dose of T3 treated groups.
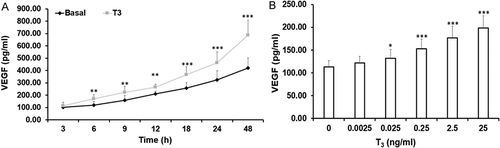
MLTC-1 cells challenged with different doses of T3 ranging from 0.0025 ng/ml to 25 ng/ml increased VEGF secretion in a dose dependent manner (). However, the increase in VEGF secretion between 2.5 ng/ml and 25 ng/ml was not significantly different. Therefore, an optimal dose of 2.5 ng/ml of T3 was selected for further experiments. The MTT assay results showed that this dose was not toxic to the MLTC-1 cells (data not shown).
Regulation of VEGF mRNA and secretion by T3
The effect of 2.5 ng/ml of T3 on VEGF mRNA levels and protein secretion by MLTC-1 cells were studied. The effect of T3 was compared to that of LH and hypoxia, the prime regulators of Leydig cell function in vivo. To reproduce the effect of LH, 8-Br-cAMP, a cAMP analogue and a second messenger of LH was used. CoCl2 was used as a chemical hypoxic mimetic.
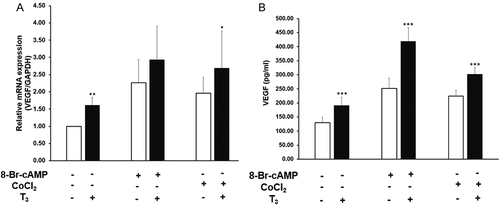
T3 significantly increased VEGF mRNA levels by 1.6 fold, and VEGF secretion by 1.5 fold (). 8-Br-cAMP (1 mM) increased VEGF mRNA levels by 2.3 fold and VEGF secretion by 1.9 fold (). The combination of T3 with 8-Br-cAMP increased VEGF mRNA by 2.9 fold and VEGF secretion by 3.2 fold (). CoCl2 (100 µM) increased VEGF mRNA levels by 2.0 fold, and VEGF secretion by 1.8 fold (). The combination of T3 with CoCl2 increased VEGF mRNA by 2.7 fold and VEGF secretion by 2.4 fold (). When T3 was combined with either 8-Br-cAMP or CoCl2, they showed an additive effect on VEGF mRNA levels and protein secretion. The stimulation of VEGF by T3 alone was significantly less than that by 8-Br-cAMP or CoCl2 alone.
Figure 2. Effect of T3 on basal, 8-Br-cAMP, and CoCl2 induced VEGF expression and secretion. (A) VEGF mRNA expression; (B) VEGF secretion. MLTC-1 cells were treated with 2.5 ng/ml of T3, 1M of 8-Br-cAMP, 100µM of CoCl2, or a combination of these as indicated for 6 hours. Bars represent mean ± SD of three experiments; each experiment was done in triplicate (n = 9). * p < 0.05; ** p < 0.01; *** p < 0.001 between without and with T3 treated groups.
Mediatory role of steroids in T3 stimulated VEGF secretion
T3 and 8-Br-cAMP were shown to stimulate steroid secretion and CoCl2-induced hypoxia was shown to inhibit steroid secretion in Leydig cell derived lines (Manna et al. Citation1999, Citation2001; Kumar et al. Citation2014). In this study also, T3 stimulated steroid secretion from MLTC-1 cells (. Studies have also shown a stimulatory effect of steroids on VEGF expression and secretion in Leydig cell derived MA-10 cells (Schwarzenbach et al. Citation2004). Therefore, we investigated whether the rise in steroids in response to T3 mediates the increase in VEGF production. To study the mediatory role of steroids in T3 stimulated VEGF production, ketoconazole, an inhibitor of steroid synthesis was used (Chang and Fung Citation2002) at a dose of 20 µM of ketoconazole. This dose was not toxic to the MLTC-1 cells as seen by the MTT assay (data not shown). Ketoconazole significantly inhibited basal, T3, 8-Br-cAMP, and CoCl2 treated progesterone secretion by 36%, 45%, 47%, and 39%, respectively (). Ketoconazole also significantly inhibited testosterone secretion by 37%, 62%, 87%, and 43%, respectively from basal, T3, 8-Br-cAMP, and CoCl2 treated cells (). For T3 and 8-Br-cAMP treated cells, progesterone secretion was partially inhibited by ketoconazole while testosterone secretion was completely abolished.
Figure 3. Effect of steroid synthesis inhibitor, ketoconazole, on secretion of steroids and VEGF by MLTC-1 cells under basal and stimulated conditions. Treatment with 20µM ketoconazole inhibited secretion of progesterone (A), testosterone (B), and VEGF (C,D) under basal and stimulated conditions. Addition of progestin R5020 (Prog) (C) and dihydrotestosterone (DHT) (D) reversed the inhibitory effect of ketoconazole on VEGF secretion. Bars represent mean ± SD of three experiments, each experiment performed in triplicate (n = 9). * p < 0.05; ** p < 0.01; *** p < 0.001 in cells treated without and with ketoconazole in the corresponding groups. ## p < 0.01; ### p < 0.001 in cells treated with only stimuli and stimuli + ketoconazole + exogenous progestin group.
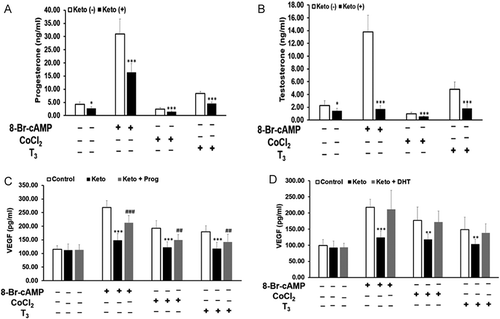
Ketoconazole showed no significant effect on the basal secretion of VEGF. Ketoconazole significantly inhibited T3, 8-Br-cAMP, and CoCl2 stimulated VEGF secretion by 30%, 47%, and 37%, respectively (). Inhibition of endogenous steroidogenesis by ketoconazole resulted in a decrease in T3 stimulated VEGF secretion suggesting a mediatory role of steroids.
To investigate whether the ketoconazole inhibited VEGF secretion was a consequence of inhibition of steroidogenesis and not due to any other independent property of ketoconazole, we used exogenous steroids, namely, non-metabolizable progestin R5020 and DHT. The ketoconazole-treated cells were incubated with 35 ng/ml of R5020 or 15 ng/ml of DHT. These doses were chosen based on the amount of progesterone and testosterone secreted by MLTC-1 cells in response to 1mM 8-Br-cAMP. When progestin R5020 was added to the ketoconazole-treated cells, VEGF secretion in response to T3, 8-Br-cAMP, and CoCl2 increased by 40%, 54%, and 38%, respectively (). Non-metabolizable progestin R5020 partially reversed the inhibition of VEGF secretion by ketoconazole. In comparison, when DHT was added to ketoconazole-treated cells, VEGF secretion in response to T3, 8-Br-cAMP, and CoCl2 increased by 77%, 93%, and 91%, respectively (). This showed that exogenous DHT treatment completely reversed the inhibition of VEGF secretion by ketoconazole.
Mediatory role of HIF-1α in T3 stimulated VEGF secretion
T3 was shown to stimulate the HIF-1α protein in a variety of cell types (Ma et al. Citation2004; Otto and Fandrey Citation2008). However, the role of T3 on HIF-1α in Leydig cells was not known. Therefore, we studied the role of T3 on HIF-1α mRNA and protein levels in MLTC-1 cells. Moreover, it was also of interest to study the mediatory role of HIF-1α in T3 induced VEGF expression and secretion in MLTC-1 cells.
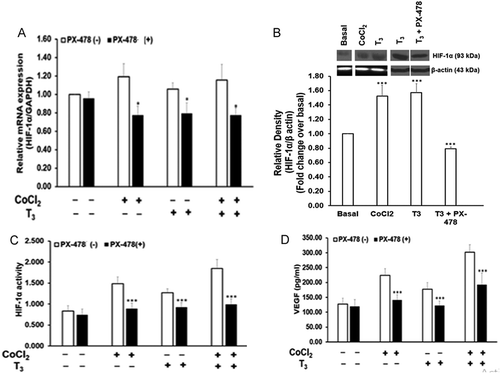
HIF-1α mRNA levels showed no significant change in response to T3 or CoCl2 in MLTC-1 cells (). However, HIF-1α protein increased 1.6 fold by T3 and 1.5 fold by CoCl2 (). HIF-1α activity increased 1.7 fold by T3 and 1.8 fold by CoCl2 ().
Figure 4. Effect of 25µM PX-478 on HIF-1α levels, activity and VEGF secretion by MLTC-1 cells under basal, CoCl2, and T3 – treated conditions. MLTC-1 cells were treated with PX-478 in the presence of 2.5 ng/ml of T3 or 100µM CoCl2 or their combination for 6 hours. HIF-1α mRNA (A), HIF-1α protein (B), HIF-1α activity (C), and VEGF secretion (D) were measured. Bars represent mean ± SD of three experiments; each experiment was done in triplicate (n = 9). * p < 0.05; *** p < 0.001 in cells treated without and with PX-478 in the corresponding groups.
To confirm the mediatory role of HIF-1α on T3 induced VEGF, PX-478, a specific inhibitor of HIF-1α, was used. A dose of 25 µM of PX-478 was selected based on previous studies (Koh et al. Citation2008). This dose of PX-478 was not toxic to the MLTC-1 cells (data not shown).
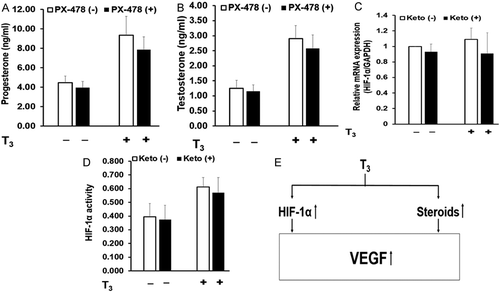
PX-478 showed no significant effect on the basal HIF-1α mRNA levels and its activity. PX-478 significantly inhibited T3- and CoCl2-stimulated HIF-1α mRNA levels by 25% and 35%, respectively (). It also inhibited T3 stimulated HIF-1α protein levels by 50% (). HIF-1α activity was also inhibited 27% and 40%, respectively by PX-478 in T3- and CoCl2-stimulated MLTC-1 cells ().
PX-478 showed no significant effect on the basal secretion of VEGF but inhibited T3- and CoCl2-stimulated VEGF secretion by 31% and 37%, respectively (). Inhibition of HIF-1α by PX-478 resulted in a decrease in T3 stimulated VEGF secretion suggesting a mediatory role of HIF-1α.
Cross-talk of steroid and HIF-1α in T3 stimulated VEGF secretion
Our results showed that T3 stimulated VEGF secretion was mediated through steroids and HIF-1α. We next investigated whether steroids and HIF-1α cross-talk with each other to increase VEGF secretion in response to T3 or form two independent pathways. To clarify this, we studied the effect of PX-478 (inhibitor of HIF-1α) on steroid secretion and the effect of ketoconazole (steroidogenesis inhibitor) on HIF-1α.
PX-478 showed no significant effect on T3 induced steroid secretion (). This suggested that regulation of T3 stimulated steroids was not mediated through HIF-1α. Similarly, ketoconazole showed no significant effect on HIF-1α mRNA levels or HIF-1α protein activity in T3 treated cells (). This suggested that regulation of T3 stimulated HIF-1α was not mediated through steroids. Thus, we showed that steroids and HIF-1α independently stimulate VEGF secretion in response to T3.
Figure 5. Independent role of steroids and HIF-1α in T3 stimulated VEGF secretion. MLTC-1 cells under basal and T3-stimulated conditions were treated with 25µM of PX-478, a HIF-1α inhibitor, and secretion of progesterone (A) and testosterone (B) was measured. Similarly, effect of 20µM of ketoconazole, a steroid synthesis inhibitor, on HIF-1α mRNA (C) and HIF-1α activity (D) was measured in MLTC-1 cells under basal and T3-stimulated conditions. Bars represent mean ± SD of three experiments; each experiment was done in triplicate. (n = 9). Based on these results, a mechanism for induction of VEGF expression and secretion in MLTC-1 cells by T3 is proposed (E).
Discussion
Regulation of VEGF by T3
In this study, we showed for the first time that T3 stimulates VEGF production in the Leydig cells. The increase in VEGF secretion in response to T3 is accompanied by a parallel increase in the VEGF mRNA levels suggesting de novo synthesis of VEGF (). Similar stimulation of VEGF by T3 has been observed in other cell types like rat brain-derived endothelial cells and human hepatoma cells (Zhang et al. Citation2010; Chen et al. Citation2014). In our study, the increase in VEGF secretion was less than two fold by all three stimulators independently, but we think the increases are physiologically relevant since hormones and growth factors act through signalling mechanisms/second messengers like cAMP, kinases, etc. Once these mechanisms are activated, they can amplify the effect of the biological signal. Moreover, T3 enhanced the action of the maximal dose of 8-Br-cAMP and CoCl2 on VEGF production (). This suggested that the presence of T3 could be a constitutive necessity for LH/cAMP and hypoxia to maximize their effect on VEGF secretion.
Steroids and HIF-1α are two well-established stimulators of VEGF secretion (Forsythe et al. Citation1996; Schwarzenbach et al. Citation2004; Eisermann et al. Citation2013). Hence, we next investigated the possibility that T3-stimulated VEGF secretion is mediated by steroids and/or HIF-1α.
Mediatory role of steroids in T3-stimulated VEGF secretion
Previous studies as well as this study showed that T3 significantly stimulates steroid secretion by primary Leydig cells and their derived lines (Manna et al. Citation1999, Citation2001; Maran et al. Citation2000). In a variety of cells such as in prostatic cells (Levine et al. Citation1998; Richard et al. Citation2002), uterine cells (Hyder et al. Citation1996; Classen-Linke et al. Citation2000), and breast cancer cells (Hyder et al. Citation1998) steroids such as androgens, estrogens, or progestin were shown to stimulate VEGF production. In MA-10 Leydig cells exogenous progesterone was shown to increase VEGF mRNA expression and protein production (Schwarzenbach et al. Citation2004). Three androgen receptor binding sites have been identified within the VEGF promoter in prostate cancer cells (Eisermann et al. Citation2013). In these prostate cell lines, two transcription factors, specificity protein-1 (Sp1) and Wilms Tumour Protein (WT1) form complexes with androgen bound androgen receptor; the complex then binds to the VEGF core promoter region. Mutation of a critical Sp1 binding site (Sp1.4) within the VEGF core promoter region abolished the increase in VEGF mRNA levels by androgen (Eisermann et al. Citation2013).
To investigate the role of steroids in T3 induced VEGF, MLTC-1 cells were treated with ketoconazole, an inhibitor of steroidogenesis. In rat testis, ketoconazole inhibited CYP17A1 and 17, 20 lyase enzyme but showed no effect on 3β-hydroxy steroid dehydrogenase (3β-HSD) (Ayub and Levell Citation1987). In MA-10 Leydig cells, ketoconazole was shown to inhibit CYP11A1 enzyme and subsequently decrease hCG- and dbcAMP-stimulated progesterone secretion (Chaudhary and Stocco Citation1989).
Inhibition of endogenous steroidogenesis by ketoconazole abolished T3, 8-Br-cAMP, and CoCl2 stimulated VEGF protein secretion (). However, it showed no significant effect on the basal secretion of VEGF. These results suggested that the increase in VEGF secretion by T3 was mediated through steroids.
To further confirm that the ketoconazole-inhibited VEGF secretion is a consequence of inhibition of steroidogenesis and not due to any other independent property of ketoconazole, exogenous steroids were added to the cells. Exogenous non-metabolizable progestin R5020 blocked the inhibitory effect of ketoconazole on VEGF secretion, though the block was not complete (). Exogenous DHT completely blocked the inhibitory effect of ketoconazole on VEGF secretion (). The fact that exogenous DHT was more effective than progestin R5020 in over-riding the inhibitory effect of ketoconazole on VEGF secretion, suggests that it is primarily the testosterone which regulates VEGF secretion and progesterone probably regulates VEGF secretion by converting to testosterone. However, in our study we used progestin R5020, which cannot metabolize to testosterone. Hence, the partial blockage of the inhibitory effect of ketoconazole on VEGF secretion by non-metabolizable progestin R5020 suggests some direct role of progesterone itself on VEGF secretion. A similar stimulation of VEGF secretion by exogenous progesterone was also reported in MA-10 Leydig cells (Schwarzenbach et al. Citation2004). MLTC-1 cells lack a classical nuclear progesterone receptor; though a non-classical receptor for progesterone has been identified in these cells (El-Hefnawy et al. Citation2000). The effect of progestin R5020 is possibly mediated through the non-classical progesterone receptor. However, in human prostate tumour cell line LNCaP, R5020 was shown to bind to androgen receptors with low affinity (10% of DHT) (Veldscholte et al. Citation1990). Hence, the effect of progestin R5020 on VEGF could also be due to its binding to the androgen receptor.
Mediatory role of HIF-1α in T3-stimulated VEGF secretion
Testicular oxygen tension has been shown to be about two-fold lower than that in core body tissues (Klotz et al. Citation1996; Carlsson et al. Citation2001; Yu and Cringle Citation2006). Low oxygen tension stabilizes the HIF-1α protein. Previous studies in primary Leydig cells and the TM3 cell line have reported a constitutive expression of HIF-1α mRNA and protein (Lysiak et al. Citation2009). Our results also showed that HIF-1α was constitutively expressed in MLTC-1 cells (). The constitutive expression of HIF-1α in Leydig cells may be an adaptive response of these cells to the physiologically low testicular oxygen tension.
Thyroid hormones increase cellular consumption of oxygen (Sterling et al. Citation1980). The T3-stimulated oxygen consumption may lead to enhanced hypoxic conditions in the testicular cells. T3 treatment of rats was shown to increase oxygen consumption by their excised testis in vitro (Zamoner et al. Citation2007) whereas propyl thiouracil (PTU) treatment reduced oxygen consumption (Zamoner et al. Citation2008). The lowering of oxygen tension by T3 could stabilize the HIF-1α protein resulting in increased accumulation of the protein in the cell. However, oxygen consumption in human hepatoma cell line, HepG2 cells under the influence of T3 showed no increase (Fandrey et al. Citation1994). In HepG2 cells, T3 was shown to stimulate only the HIF-1α protein synthesis without affecting the stability of the HIF-1α protein or its mRNA levels (Ma et al. Citation2004).
T3 was shown to stimulate the HIF-1α protein in a variety of cell types (Ma et al. Citation2004; Otto and Fandrey Citation2008). In our study, though T3 did not change the HIF-1α mRNA levels in MLTC-1 cells (), it increased HIF-1α protein levels and its activity (). The increase in HIF-1α protein levels could be due to two reasons, inhibition of protein degradation and/or increase in the protein synthesis. However, which possibility holds true in Leydig cells needs to be further investigated.
Hypoxia and hypoxia – induced HIF-1α are well-established upstream regulators of VEGF. HIF-1α binds to the VEGF gene enhancer region and stimulates its transcription (Forsythe et al. Citation1996). In MA-10 cells, the increase in VEGF secretion in response to CoCl2 was mediated through HIF-1α (Kumar et al. Citation2012).
To establish a role of HIF-1α in T3-stimulated VEGF secretion, we used PX-478, a HIF-1α inhibitor. PX-478 was shown to inhibit transcription and translation of HIF-1α in different cancer cell lines (Koh et al. Citation2008). The same study also showed that PX-478 inhibited VEGF secretion (Koh et al. Citation2008). Our results in MLTC-1 cells showed that PX-478 did not affect basal HIF-1α mRNA levels or its activity. PX-478 reduced T3 treated HIF-1α mRNA and protein levels as well as its activity (). PX-478 abolished T3-stimulated VEGF secretion though it had no effect on the basal VEGF secretion (). These observations suggest a mediatory role of HIF-1α in T3-induced VEGF secretion.
Steroids and HIF-1α mediate T3-stimulated VEGF secretion independent of each other
In the current study, ketoconazole, a specific inhibitor of steroidogenesis, did not significantly affect HIF-1α mRNA levels or its activity in T3 treated cells (). Also, the specific HIF-1α inhibitor, PX-478, showed no effect on T3-stimulated steroid secretion (). These results suggested that T3-stimulated VEGF secretion was mediated by steroids and HIF-1α independently. In contrast, a stimulatory role of DHT on the HIF-1α protein has been shown in prostate cancer cell line, LNCaP (Mabjeesh et al. Citation2003). Our earlier study on MA-10 cells showed that the inhibitory effect of CoCl2 on progesterone secretion was mediated partly through HIF-1α (Kumar et al. Citation2014). This could have been because cyclosporine A (CsA), a non-specific inhibitor of HIF-1α with many other functions, was used in that study.
In conclusion, our results clearly demonstrate for the first time that T3 stimulates the expression and secretion of VEGF by Leydig cells. This effect of T3 is indirectly mediated by an increase in steroid secretion and HIF-1α protein levels which independently stimulate VEGF secretion. However, T3 seems to be a constitutive necessity for both cAMP and hypoxia to maximize their effect on VEGF. summarizes the mechanisms involved in T3 induced VEGF secretion by the MLTC-1 Leydig cells.
Limitations of the study
Since Leydig cells comprise <1% of total cells of the testis, it is difficult to obtain large quantities of highly viable Leydig cells and even a multi-step procedure cannot yield 100% purified Leydig cells (Salva et al. Citation2001). The present study required a large number of cells; hence, we chose MLTC-1 cells which behave physiologically in many ways and are a good model for studying Leydig cell biology. They express specific cell surface receptors for hCG which are coupled to adenylate cyclase; they express StAR and CYP17A1; they produce both progesterone and testosterone. Previous studies (Manna et al. Citation1999, Citation2001; Maran et al. Citation2000) have shown functional thyroid hormone receptor (TR) in MLTC-1 cells. They also showed that T3 stimulates StAR gene expression and steroid production in MLTC-1 cells similar to primary Leydig cells. However, future studies should be done on primary Leydig cells to show that similar effects are observed in those cells and the effects are not due to the MLTC-1 cell line being immortalized.
The doses of the various inhibitors used in this study were tested for toxicity using the MTT assay and were found to be non-toxic at the concentrations used. However, it is possible that these doses may be toxic to primary Leydig cells though they showed no toxicity on MLTC-1 cells.
VEGF exists in different isoforms which have angiogenic (VEGF164) or anti-angiogenic (VEGF165b) properties and each exerts opposing effects on spermatogenesis (Caires et al. Citation2012; Sargent et al. Citation2016). However, the current study was not designed to delineate which isoform of VEGF is up-regulated by T3 hormone in Leydig cells. This should be addressed in future studies.
Materials and methods
Cell culture
MLTC-1 cells purchased from ATCC (Manassas, USA) were maintained in Waymouth MB 752/1 medium (Sigma Aldrich, St. Louis, MO, USA) supplemented with 10% heat-inactivated fetal bovine serum (Sigma Aldrich) at 37°C in 5% CO2 in a humidified incubator. All experiments for this study were conducted on MLTC-1 cells between passage numbers 10–30.
The cells were seeded at a density of 2 × 105 cells/ml/well for estimating VEGF and steroid secretion and at 1 × 106 cells/ml/well for total RNA extraction, Western blot, and HIF-1α activity assay. Cells were treated with varying concentrations of T3, 1mM of 8-Br-cAMP, 100 µM CoCl2, or their combinations. Viability of the cells after treatment was measured using [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] (MTT, Sigma Aldrich). In experiments with HIF-1α and steroid inhibitors, cells were pre-incubated separately for 1 h with 25 µM PX-478 (S-2-amino-3-[4ʹ-N, N,-bis (chloroethyl) amino] phenyl propionic acid N-oxide dihydrochloride) or 20 µM ketoconazole, respectively. Ketoconazole and PX-478 were procured from Sigma Aldrich company.
VEGF ELISA
VEGF secretion was measured in the cell supernatant using Mouse Quantikine VEGF ELISA kit from R&D Systems (Minneapolis, MN, USA) according to the manufacturer’s instructions. This kit specifically measures mouse VEGF164 protein.
Radioimmunoassay
Progesterone and testosterone secretion was measured in cell supernatant by radioimmunoassay (RIA) according to the WHO guidelines (Sufi et al. Citation1993). Tritium labelled radioactive progesterone and testosterone were obtained from Perkin Elmer (Waltham, MA, USA). Mouse monoclonal testosterone antibody (clone 4E1G2) and rabbit polyclonal progesterone antibody were procured from Bio-Rad (Hercules, CA, USA) and used at a dilution of 1:500. The progesterone antibody used for RIA was highly specific and showed no cross-reactivity for pregnenolone or for testosterone. However, the testosterone antibody also recognizes 5α-dihydrotestosterone (DHT, 1%) and androstenedione (1%). The intra-assay variation was less than 5% and the inter-assay variation was less than 10%.
RNA extraction and quantitative real-time PCR
Total RNA was extracted using Ribozol RNA extraction reagent (Amresco, Solon, OH, USA) as per manufacturer’s instructions. Total RNA isolated from the cells was reverse transcribed using oligo dT primers and RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). The cDNA synthesized was amplified using gene specific primers (Sigma Aldrich). The sequences of VEGF primers were 5ʹ-GCACCCACGACAGAAGGGGAG-3ʹ and 5ʹ-TCACCGCCTTGGCTTGTCACA-3ʹ (Anand et al. Citation2003). The sequences of HIF-1α primers were 5ʹ-TGCTTGGTGCTGATTTGTGA-3ʹ and 5ʹ-GGTCAGATGATCAGAGTCCA-3ʹ (Kumar et al. Citation2012). The sequences of GAPDH primers were 5ʹ-ACGGGAAGCTTGTCATCAAT-3ʹ and 5ʹ-TGGACTCCACGACGTCGTACTCA-3ʹ (Konieczna et al. Citation2015). The PCR conditions used were initial denaturation at 95°C for 5 min followed by 40 cycles of 95°C for 20 s, 56°C for 20 s, and 72°C for 20 s. The qPCR was performed on Bioer LineGene 9600 thermocycler (Bioer, China) and its software was used to generate Ct values. Ct values obtained during the real time PCR reaction were used to calculate the relative mRNA expression using the formula:
Relative mRNA expressionVEGF or HIF-1α = 2−∆∆Ct
where ∆Ct = CtVEGF or HIF-1α – CtGAPDH and ∆∆Ct = ∆CtBasal – ∆CtTreated.
Western blot
Fifty µg of total protein extract was separated by SDS-PAGE using 10% resolving gel. The protein bands in the gel were transferred onto a 0.45µm PVDF membrane. PVDF membrane was blocked with 5% skimmed milk solution made in TBS-T (0.1% Tween-20 in 1X TBS). The membrane was probed with rabbit HIF-1α antibody at 1:500 dilution (Abcam, Cambridge, UK) overnight at 4°C. Anti-rabbit HRP labelled secondary antibody (CST, Boston, MA, USA) was used at a dilution of 1:3000. Protein bands were visualized using ECL system (Merck Millipore, Billerica, MA, USA). The density of the protein bands was measured using ImageJ software developed at the National Institutes of Health (Bethesda, MD, USA).
HIF-1α activity assay
MLTC-1 cells were lyzed with 1ml of freshly prepared lysis buffer A (10 mM HEPES, pH 7.9; 1.5 M MgCl2; 10 mM KCl; 0.5 mM dithiothreitol; 0.1% NP-40; 2 mM Na3VO4; 5 mM NaF; 3 µg/ml aprotinin; 25 µg/ml leupeptin; 25 µg/ml pepstatin; 25 µg/ml chymostatin; 0.2 mM PMSF) followed by centrifugation at 16,000g for 5 min at 4ºC. Cytosolic supernatant was removed and the nuclear pellet was solubilized in 10 µl of freshly prepared lysis buffer B (20 mM HEPES; 1.5 M MgCl2; 420 mM NaCl; 0.5 mM dithiothreitol; 25% glycerol; 2 mM Na3VO4; 5 mM NaF; 3 µg/ml aprotinin; 25 µg/ml leupeptin; 25 µg/ml chymostatin; 0.2 mM PMSF). It was incubated on ice for 20 min and then centrifuged for 5 min at 16,000g at 4ºC. The protein concentration in the supernatant (nuclear extract) was measured by Bradford’s reagent (Sigma Aldrich). HIF-1α activity was measured using a kit from R&D Systems according to manufacturer’s instructions. An immobilized capture antibody specifically binds HIF-1α. After washing away unbound material, a biotinylated detection antibody specific for HIF-1α was used to detect the protein, utilizing a standard Streptavidin-HRP format.
Statistical analysis
All the treatments were carried out in triplicate and each experiment was repeated at least three times. The data were pooled and analyzed by using GraphPad Prism 4.0 software (GraphPad Prism Software Inc., San Diego, CA, USA). Student’s t test/ANOVA was used to calculate the p-value. p < 0.05 was considered significant.
Ethical clearance
The study was approved by the Institute Ethics Committee (Ref. No. IEC/NP-02).
Acknowledgments
Authors are thankful to Kishore Pant and Parul Gupta, Ph.D. Students, Department of Life Sciences and Biotechnology, South Asian University for their help in the Western blot experiments. Authors are also thankful to M. Marim, Medical Lab technologist and N. Tyagi, Technical Officer, Department of Reproductive Biology, All India Institute of Medical Sciences for their help in performing some of the experiments.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Notes on contributors
Anand Kumar
Carried out the experiments, analyzed the experimental results, and wrote the manuscript: BD;
Designed some of the experiments, analyzed the experimental results, and wrote the manuscript: SG;
Carried out the western blot experiments: SKV;
Conceptualized the study, designed the experiments and checked the manuscript: AK.
References
- Anand RJ, Paust HJ, Altenpohl K, Mukhopadhyay AK. 2003. Regulation of vascular endothelial growth factor production by Leydig cells in vitro: the role of protein kinase A and mitogen-activated protein kinase cascade. Biol Reprod. 68(5):1663–1673.
- Ayub M, Levell MJ. 1987. Inhibition of testicular 17 alpha-hydroxylase and 17,20-lyase but not 3 beta-hydroxysteroid dehydrogenase-isomerase or 17 beta-hydroxysteroid oxidoreductase by ketoconazole and other imidazole drugs. J Steroid Biochem. 28(5):521–531.
- Caires KC, De Avila JM, Cupp AS, McLean DJ. 2012. VEGFA family isoforms regulate spermatogonial stem cell homeostasis in vivo. Endocrinology. 153(2):887–900.
- Carlsson PO, Palm F, Andersson A, Liss P. 2001. Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes. 50(3):489–495.
- Chang CL, Fung HP. 2002. Effects of ketoconazole on progesterone and cAMP production in MA-10 mouse Leydig tumor cells. Biol Pharm Bull. 25(6):794–797.
- Chaudhary LR, Stocco DM. 1989. Inhibition of hCG- and cAMP-stimulated progesterone production in MA-10 mouse Leydig tumor cells by ketoconazole. Biochem Int. 18(1):251–262.
- Chen CY, Chung IH, Tsai MM, Tseng YH, Chi HC, Tsai CY, Lin YH, Wang YC, Chen CP, Wu TI, et al. 2014. Thyroid hormone enhanced human hepatoma cell motility involves brain-specific serine protease 4 activation via ERK signaling. Mol Cancer. 13:162.
- Classen-Linke I, Alfer J, Krusche CA, Chwalisz K, Rath W, Beier HM. 2000. Progestins, progesterone receptor modulators, and progesterone antagonists change VEGF release of endometrial cells in culture. Steroids. 65(10–11):763–771.
- Eisermann K, Broderick CJ, Bazarov A, Moazam MM, Fraizer GC. 2013. Androgen up-regulates vascular endothelial growth factor expression in prostate cancer cells via an Sp1 binding site. Mol Cancer. 12:7.
- El-Hefnawy T, Manna PR, Luconi M, Baldi E, Slotte JP, Huhtaniemi I. 2000. Progesterone action in a murine Leydig tumor cell line (mLTC-1), possibly through a nonclassical receptor type. Endocrinology. 141(1):247–255.
- Fandrey J, Pagel H, Frede S, Wolff M, Jelkmann W. 1994. Thyroid hormones enhance hypoxia-induced erythropoietin production in vitro. Exp Hematol. 22(3):272–277.
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. 1996. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 16(9):4604–4613.
- Hwang GS, Wang SW, Tseng WM, Yu CH, Wang PS. 2007. Effect of hypoxia on the release of vascular endothelial growth factor and testosterone in mouse TM3 Leydig cells. Am J Physiol Endocrinol Metab. 292(6):E1763–E1769.
- Hyder SM, Stancel GM, Chiappetta C, Murthy L, Boettger-Tong HL, Makela S. 1996. Uterine expression of vascular endothelial growth factor is increased by estradiol and tamoxifen. Cancer Res. 56(17):3954–3960.
- Hyder SM, Murthy L, Stancel GM. 1998. Progestin regulation of vascular endothelial growth factor in human breast cancer cells. Cancer Res. 58(3):392–395.
- Iitaka M, Miura S, Yamanaka K, Kawasaki S, Kitahama S, Kawakami Y, Kakinuma S, Oosuga I, Wada S, Katayama S. 1998. Increased serum vascular endothelial growth factor levels and intrathyroidal vascular area in patients with Graves’ disease and Hashimoto’s thyroiditis. J Clin Endocrinol Metab. 83(11):3908–3912.
- Jana NR, Bhattacharya S. 1994. Binding of thyroid hormone to the goat testicular Leydig cell induces the generation of a proteinaceous factor which stimulates androgen release. J Endocrinol. 143(3):549–556.
- Klotz T, Vorreuther R, Heidenreich A, Zumbé J, Engelmann U. 1996. Testicular tissue oxygen pressure. J Urol. 155(4):1488–1491.
- Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, Powis G. 2008. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther. 7(1):90–100.
- Konieczna A, Szczepańska A, Sawiuk K, Węgrzyn G, Łyżeń R. 2015. Effects of partial silencing of genes coding for enzymes involved in glycolysis and tricarboxylic acid cycle on the enterance of human fibroblasts to the S phase. BMC Cell Biol. 16:16.
- Kumar A, Rani L, Dhole B, Chaturvedi PK. 2012. Oxygen as a regulator of MA-10 cell functions: effect of cobalt chloride on vascular endothelial growth factor production. Andrologia. 44(Suppl 1):615–620.
- Kumar A, Rani L, Dhole B. 2014. Role of oxygen in the regulation of Leydig tumor derived MA-10 cell steroid production: the effect of cobalt chloride. Syst Biol Reprod Med. 60(2):112–118.
- Levine AC, Liu XH, Greenberg PD, Eliashvili M, Schiff JD, Aaronson SA, Holland JF, Kirschenbaum A. 1998. Androgens induce the expression of vascular endothelial growth factor in human fetal prostatic fibroblasts. Endocrinology. 139(11):4672–4678.
- Lysiak JJ, Kirby JL, Tremblay JJ, Woodson RI, Reardon MA, Palmer LA, Turner TT. 2009. Hypoxia-inducible factor-1alpha is constitutively expressed in murine Leydig cells and regulates 3beta-hydroxysteroid dehydrogenase type 1 promoter activity. J Androl. 30(2):146–156.
- Ma Y, Freitag P, Zhou J, Brüne B, Frede S, Fandrey J. 2004. Thyroid hormone induces erythropoietin gene expression through augmented accumulation of hypoxia-inducible factor-1. Am J Physiol Regul Integr Comp Physiol. 287(3):R600–R607.
- Mabjeesh NJ, Willard MT, Frederickson CE, Zhong H, Simons JW. 2003. Androgens stimulate hypoxia-inducible factor 1 activation via autocrine loop of tyrosine kinase receptor/phosphatidylinositol 3’-kinase/protein kinase B in prostate cancer cells. Clin Cancer Res. 9(7):2416–2425.
- Manna PR, Tena-Sempere M, Huhtaniemi IT. 1999. Molecular mechanisms of thyroid hormone-stimulated steroidogenesis in mouse leydig tumor cells. Involvement of the steroidogenic acute regulatory (StAR) protein. J Biol Chem. 274(9):5909–5918.
- Manna PR, Kero J, Tena-Sempere M, Pakarinen P, Stocco DM, Huhtaniemi IT. 2001. Assessment of mechanisms of thyroid hormone action in mouse Leydig cells: regulation of the steroidogenic acute regulatory protein, steroidogenesis, and luteinizing hormone receptor function. Endocrinology. 142(1):319–331.
- Maran RR, Arunakaran J, Aruldhas MM. 2000. T3 directly stimulates basal and modulates LH induced testosterone and oestradiol production by rat Leydig cells in vitro. Endocr J. 47(4):417–428.
- Otto T, Fandrey J. 2008. Thyroid hormone induces hypoxia-inducible factor 1alpha gene expression through thyroid hormone receptor beta/retinoid x receptor alpha-dependent activation of hepatic leukemia factor. Endocrinology. 149(5):2241–2250.
- Richard C, Kim G, Koikawa Y, Salm SN, Tsujimura A, Wilson EL, Moscatelli D. 2002. Androgens modulate the balance between VEGF and angiopoietin expression in prostate epithelial and smooth muscle cells. Prostate. 50(2):83–91.
- Salva A, Klinefelter GR, Hardy MP. 2001. Purification of rat leydig cells: increased yields after unit-gravity sedimentation of collagenase-dispersed interstitial cells. J Androl. 22(4):665–671.
- Sargent KM, Clopton DT, Lu N, Pohlmeier WE, Cupp AS. 2016. VEGFA splicing: divergent isoforms regulate spermatogonial stem cell maintenance. Cell Tissue Res. 363(1):31–45.
- Schwarzenbach H, Chakrabarti G, Paust HJ, Mukhopadhyay AK. 2004. Gonadotropin-mediated regulation of the murine VEGF expression in MA-10 Leydig cells. J Androl. 25(1):128–139.
- Sterling K, Brenner MA, Sakurada T. 1980. Rapid effect of triiodothyronine on the mitochondrial pathway in rat liver in vivo. Science. 210(4467):340–342.
- Sufi SB, Donaldson A, Jeffcoate SL. 1993. Programme for the provision of matched assay reagents for the radioimmunoassay of hormones in reproductive physiology. In: Method manual. Geneva, Switzerland: World Health Organization.
- Veldscholte J, Voorhorst-Ogink MM, Bolt-De Vries J, Van Rooij HC, Trapman J, Mulder E. 1990. Unusual specificity of the androgen receptor in the human prostate tumor cell line LNCaP: high affinity for progestagenic and estrogenic steroids. Biochim Biophys Acta. 1052(1):187–194.
- Yu DY, Cringle SJ. 2006. Oxygen distribution in the mouse retina. Invest Ophthalmol Vis Sci. 47(3):1109–1112.
- Zamoner A, Barreto KP, Filho DW, Sell F, Woehl VM, Guma FC, Silva FR, Pessoa-Pureur R. 2007. Hyperthyroidism in the developing rat testis is associated with oxidative stress and hyperphosphorylated vimentin accumulation. Mol Cell Endocrinol. 267(1–2):116–126.
- Zamoner A, Barreto KP, Filho DW, Sell F, Woehl VM, Guma FC, Pessoa-Pureur R, Silva FR. 2008. Propylthiouracil-induced congenital hypothyroidism upregulates vimentin phosphorylation and depletes antioxidant defenses in immature rat testis. J Mol Endocrinol. 40(3):125–135.
- Zhang L, Cooper-Kuhn CM, Nannmark U, Blomgren K, Kuhn HG. 2010. Stimulatory effects of thyroid hormone on brain angiogenesis in vivo and in vitro. J Cereb Blood Flow Metab. 30(2):323–335.