
ABSTRACT
Marfan syndrome (MFS), an autosomal dominant heritable disease of the connective tissue, is characterized by broad clinical manifestations in the musculoskeletal, cardiovascular, pulmonary, and ocular systems. In this study, a male patient with MFS caused by a heterozygous mutation NM_000138.5(FBN1):c.6037 + 2 T > C in the fibrillin 1 gene (FBN1) underwent preimplantation genetic testing (PGT) by using affected-embryo-based single nucleotide polymorphism (SNP) haplotyping. Multiple displacement amplification was used for whole genome amplification of biopsied trophectoderm cells after controlled ovarian stimulation. Sanger sequencing and next-generation sequencing (NGS) were used to detect the state of FBN1 mutation. A total of 14 blastocysts formed after intracytoplasmic sperm injection were biopsied. After NGS, 60 informative polymorphic SNP markers located upstream and downstream of the FBN1 gene and its pathogenic mutation site were linked to individual alleles. Sanger sequencing further confirmed that 8 blastocysts carried the mutation NM_000138.5(FBN1):c.6037 + 2 T > C, while 6 did not. Four of the non-carriers were euploid verified by copy number variation results. A female infant without MFS was born at 37 weeks gestation after a subsequent frozen embryo transfer. In conclusion, the successful case indicates that SNP haplotyping using sibling embryos as a reference is applicable to PGT in monogenetic diseases.
Abbreviations MFS: Marfan syndrome; PGT: preimplantation genetic testing; FBN1: fibrillin 1 gene; NGS: next-generation sequencing; SNP: single nucleotide polymorphism.
Introduction
Marfan syndrome (MFS), an autosomal dominant heritable disease of the connective tissue, is characterized by broad clinical manifestations in the musculoskeletal, cardiovascular, pulmonary, and ocular systems (Keane and Pyeritz Citation2008). The estimated prevalence is approximately 1–2:10ʹ000 (Groth et al. Citation2015).
Previous studies have found that over 90% of MFS cases are caused by different heterozygous sequence variants in the fibrillin-1 gene (FBN1, OMIM: 154,700) (Loeys et al. Citation2010; Li et al. Citation2012). However, more than 25% of the cases are sporadic due to de novo mutations and without a dominant family history trait (Dietz and Pyeritz Citation1995; Xiao et al. Citation2017). The pathogenic mutations changed the quantity or structure of FBN1 protein and resulted in wide clinical symptoms ranging from mild isolated traits to severe life-threatening multiorgan abnormalities (Dietz et al. Citation1991; Loeys et al. Citation2010; Agg et al. Citation2014). Therefore, PGT appears to be the efficient strategy to select embryos without pathogenic genetic defects for MFS patients (Yan et al. Citation2014).
Meanwhile, a small amount of DNA provided from biopsied trophectoderm cells and amplification bias based on WGA and polymerase chain reaction (PCR) can lead to allele dropout (ADO) and misdiagnosis (Wilton et al. Citation2009; Harper et al. Citation2012). To identify ADO to ensure the accuracy of PGT, SNPs linked to the mutated genes are used to establish haplotypes via NGS (Chen et al. Citation2017).
Haplotypes are usually constructed using the peripheral blood DNA information of the proband and his/her parents (Chen et al. Citation2017). Alternatively, pre-PGT single-sperm or discarded-embryo haplotyping was performed to confirm the family’s haplotypes without relatives as reference (Chen et al. Citation2019). However, all these methods would increase the cost and time for patients due to extra laboratory steps.
In this case report, we successfully conducted PGT for MFS by SNP haplotyping using sibling embryos as a reference.
Results
Patient and relatives
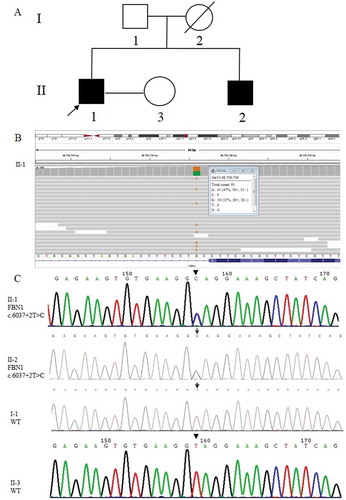
A couple, of which the male was affected by MFS, attended our reproductive medical center in 2017 for genetic consultation to prevent MFS transmission to the next generation. The 33-year-old male proband was 183 cm in height, 52 kg in weight, with a long narrow face having a mechanical heart valve replacement in 2013. The peripheral blood samples of the male proband, his wife, and the immediate family members (father and brother) were collected for pedigree analysis (). The proband’s mother (I-2) died of heart disease at the age of 42 (a suspected MFS patient). The result of NGS showed that the MFS in the proband (II-1) was caused by a heterozygous mutation NM_000138.5 (FBN1):c.6037 + 2 T > C in intron 49 of the FBN1 gene, which was also confirmed by Sanger sequencing. Pedigree analysis of this MFS family () through Sanger sequencing revealed that the proband’s younger brother (II-2) carried the same FBN1 gene mutation while his father (I-1) and his wife (II-3) were wild-type (WT). The couple underwent PGT treatment, including ovarian stimulation, oocyte retrieval, intracytoplasmic sperm injection, embryo culture, trophectoderm biopsy, blastocyst vitrification, and frozen embryo transfer (see Supplemental Material).
Figure 1. Causative variant detection in this family. (A) Pedigree of the MFS family reported in this study. Squares and circles indicate males and females, respectively. The black symbols II-1 (the proband) and II-2 (his younger brother) represent the male patients who suffered from MFS. The blank symbols show I-1 (his father) and II-3 (his wife) were unaffected and I-2 (his mother died of heart disease without specific diagnosis). (B) The pathogenic mutation site (FBN1: NM_000138; chr15:48,736,736 c.6037 + 2 T > C) of II-1 (the proband) detected by NGS. The figure provides the result of the antisense chain reads. (C) Sanger sequencing results of the FBN1 gene in the MFS family. II-1 and II-2 carried the FBN1 gene mutation c.6037 + 2 T > C, while I-1 and II-3 were wild-type (WT)
Intracytoplasmic sperm injection, embryo culture, trophectoderm biopsy, and blastocyst vitrification
After luteal phase down-regulation and controlled ovarian stimulation, 26 oocytes were obtained. Twenty-five metaphase II-stage (MII) oocytes were then inseminated by ICSI, and 23 had fertilized normally as indicated by the presence of two pronuclei. On day 3 post-insemination, 21 fertilized oocytes developed into 7–9 cells. Finally, a total of 14 blastocysts were obtained. Ten were subject to trophectoderm cell biopsy on day 5 and the other 4 on day 6 ().
Table 1. Results of preimplantation genetic testing of 14 biopsied samples
WGA and Sanger sequencing of embryos
WGA was successfully performed for all the 14 biopsied samples by MDA. The genetic mutation site was directly detected by Sanger sequencing. The results showed that 8 blastocysts carried the mutation NM_000138.5 (FBN1):c.6037 + 2 T > C ().
NGS-based SNP haplotyping and copy number variation
ADO, defined as random amplification failure of one or both alleles (Locus drop out, LDO), was observed in most samples except E7, E12, and E13. The recombination region was not detected in any of the samples. Finally, 8 blastocysts (E3-E7, E13, and E14) were confirmed to exhibit an affected haplotype pattern and carry the paternal mutation NM_000138.5 (FBN1):c.6037 + 2 T > C. In comparison, 6 blastocysts (E1, E2, and E9-E12) possessed an unaffected haplotype with the normal paternally inherited allele and did not carry the paternal mutation (, ), consistent with the preliminary results of Sanger sequencing ().
Figure 2. Haplotyping results. 60 informative polymorphic SNP markers located upstream and downstream of the pathogenic site were selected to establish the haplotype. Ten of these SNPs (rs48523284, rs48540199, rs48564160, rs48584883 and rs48633092 are located upstream of the pathogenic site; rs48967277, rs49247626, rs49290738, rs49338132 and rs49359620) are selected for this figure. Diagram showing the results of haplotype analysis of the male, female and 14 blastocysts (E1-14). E3-8 and E13-14 inherited an affected haplotype (yellow-line allele) from the male, whereas E1, E2, and E9-12 inherited a normal haplotype (orange-line allele). ‘?’ indicates ADO or LDO
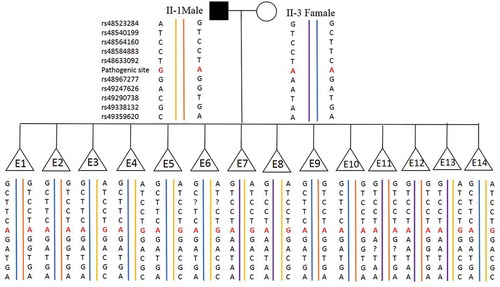
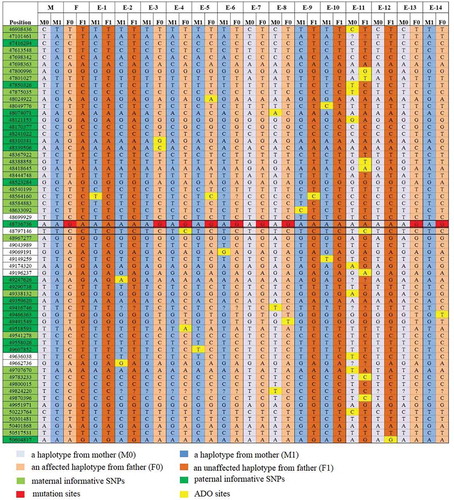
Figure 3. NGS-based SNP haplotyping results of mother (M), father (F), and 14 embryos (E1-E14). 60 informative polymorphic SNP markers located upstream and downstream of the mutation site in FBN1 were selected to establish the haplotype. The positions marked in green color are paternal informative SNPs while the positions in light green are maternal. The red squares in position 48,736,736 represent the mutation sites (G in red) and the yellow ones represent ADO sites. E1, E2, and E9-E12 exhibited an unaffected haplotype with the normal paternally inherited allele (F1), whereas E3-E7, E13, and E14 exhibited an affected haplotype pattern (F0). Question mark represents the undetected site which was actually a ‘Locus drop out’. Single solid line represents the gene boundary, while double solid line represents the pathogenic site boundary
Before embryo transfer, preimplantation genetic testing for aneuploidies (PGT-A) was carried out to detect the CNV of the 6 non-carriers chromosomes. The results indicated that E1, E2, E9, and E10 were euploid ( and ).
Figure 4. Copy number variation results of 6 samples (E1, E2, E9-12). CNV of the 6 embryos with normal results of Sanger sequencing and SNP haplotyping (E1, E2, E9-12) were shown both in and (sex chromosomes not shown), while the other 8 embryos with abnormal results of Sanger sequencing and SNP haplotyping (E3-8, E13, E14) were not tested for CNV. The maps shown that sample E1, E2, E9 and E10 were diploid, whereas E11 was mosaic [46, XX, del mosaic (2) (47.7%), del mosaic (4) (46.8%)] and E12 was aneuploid (47, XX, dup19)
![Figure 4. Copy number variation results of 6 samples (E1, E2, E9-12). CNV of the 6 embryos with normal results of Sanger sequencing and SNP haplotyping (E1, E2, E9-12) were shown both in Table 1 and Figure 3 (sex chromosomes not shown), while the other 8 embryos with abnormal results of Sanger sequencing and SNP haplotyping (E3-8, E13, E14) were not tested for CNV. The maps shown that sample E1, E2, E9 and E10 were diploid, whereas E11 was mosaic [46, XX, del mosaic (2) (47.7%), del mosaic (4) (46.8%)] and E12 was aneuploid (47, XX, dup19)](/cms/asset/caa4ee28-dd3b-45e9-87cd-bbc67b956203/iaan_a_1926574_f0004_oc.jpg)
Frozen embryo transfer results
Clinical pregnancy was defined as the presence of a fetal heartbeat by sonography 28 days after frozen embryo transfer (FET). Conventional Sanger sequencing of the fetal DNA obtained by amniocentesis at 17 weeks of gestation confirmed the diagnosis of PGT. A healthy female infant weighing 3300 grams was born at 37th week of gestation. Sanger sequencing of the newborn baby’s blood sample showed that she did not carry the pathogenic mutation.
Discussion
In the present study, MFS resulted from a heterozygous mutation NM_000138.5 (FBN1):c.6037 + 2 T > C in intron 49 of the FBN1 gene. The same causative mutation was also identified as a pathogenic mutation in a previous report (Yang et al. Citation2016).
For monogenetic diseases like MFS, Sanger sequencing as the most direct gene mutation detection method could not guarantee the accuracy of PGT because of ADO caused by preferential amplification from a small amount of DNA in blastocyst biopsy followed by PGT related technology (Shen et al. Citation2011; Chen et al. Citation2019). SNPs as polymorphic markers linked to the mutation site have been employed to analyze haplotypes to ensure the accuracy of PGT (Shen et al. Citation2011; Chen et al. Citation2019).
In this study, the haplotype was established based on NGS to verify Sanger sequencing results and avoid the misdiagnosis due to ADO (Renwick et al. Citation2010; Harper et al. Citation2012). Haplotypes are usually constructed using the information from the proband’s peripheral blood DNA and his/her parents. As reported previously, two main methods could be used to establish the haplotype for linkage analysis if the proband had no affected relatives. (1) Pre-PGT single-sperm haplotyping:: Single sperm was isolated, the genotypes of SNP alleles determined via NGS then analyzed (Chen et al. Citation2019). Because a spermatozoon is haploid, it is easy to establish haplotype from individual sperm and distinguish the inherited mutant haplotype from the normal one (Altarescu et al. Citation2006). However, the technical limitation was that the polymorphic markers had to be identified before linkage analysis and the method required multiple steps. (2) Post-ICSI discarded-embryo haplotyping: The haplotypes of the discarded embryos were retrospectively analyzed. Using discarded embryos could overcome the problem of insufficient genomic DNA in PGT, which was the key reason for ADO and misdiagnosis. However, it would require additional laboratory work increasing medical expenses.
Genetic sequencing analysis of the MFS family in our study revealed that the proband’s father did not carry the pathogenic mutation. We could not confirm whether the pathogenic mutation was inherited from the proband’s mother because she died of heart disease at the age of 42. Studies had shown that over 25% of the cases were spontaneous mutations without a positive family history (Dietz and Pyeritz Citation1995). However, the proband’s younger brother carried the same FBN1 gene mutation. Considering that the proband’s mother died of heart disease, we speculated that the pathogenic mutation most likely originated from his mother or the proband’s father had a slight possibility to be gonadal mosaicism. Since his mother’s DNA was not available, we could have established haplotype from the proband’s father and brother or perform haplotyping of single-sperm or discarded embryos. However, this time-consuming and laborious process would result in a relatively long delay and require direct family members recruitment (Zeevi et al. Citation2019).
Prior to ovarian stimulation, the proband’s wife was assessed as a hyper responder due to her age (29-year-old), high AMH level (6.43 ng/ml), and antral follicle account (>24), which indicated that sufficient blastocysts could be obtained for biopsy. Furthermore, since the risk of recurrence of the dominantly inherited disease was 50%, it was likely that some affected embryos would be available for haplotyping linkage analysis. Therefore, in the present study, affected embryos obtained from ICSI treatment were finally selected as reference for haplotype phasing to avoid the multiple steps before PGT and the possibility of gonadal mosaicism in the proband’s father. This alternative approach using sibling embryos as a reference could simplify procedures and reduce the total cost to patients.
NGS was first applied to preimplantation genetic diagnosis (PGD) in 2013 (Sueoka Citation2016). With the continuous development of NGS over the past 7 years, the cost and time required for sequencing has decreased significantly (Guo et al. Citation2018). NGS-based PGT can simultaneously detect target mutation sites and linked SNPs, making it possible to provide multiple diagnostic results with the advantages of high accuracy, throughput, and sensitivity (Chamayou et al. Citation2019).
Haplotyping linkage analysis with more informative SNPs can detect ADO, distinguish chromosomal monosomy and identify meiotic recombination. In this study, 60 SNP markers within 1Mb upstream and downstream of the pathogenic mutation site were used to establish the haplotype and distinguish the chromosome carrying the mutation. Even low ADO observed in the study did not affect the accuracy of the result. We could deduce whether the disease-carrying allele from the proband was present in each embryo by detecting these SNPs.
As an alternative, Karyomapping using Microarray-based SNP genotyping also provides a universal method for linkage-based diagnosis of monogenetic disorders (Handyside et al. Citation2010; Natesan et al. Citation2014). Karyomapping combined with whole genome amplification, and minisequencing for the mutation validates the clinical application to use sibling embryos as a reference in PGT of monogenetic diseases (Thornhill et al. Citation2015).
Karyomapping combines the detection of any monogenic disorder with comprehensive chromosome screening in a single test that requires no prior clinical workup of a specific test beforehand. However, Karyomapping cannot detect gene mutation sites directly whereas NGS can perform mutation detection and haplotype analysis simultaneously.
Using affected embryos to establish haplotyping would be limited in the following situations: (1) There were not enough informative SNPs to establish haplotyping; (2) Patient with poor ovarian response would not obtain sufficient embryos as reference; (3) ADO or recombination occurred in affected embryos would lead to misdiagnosis; (4) The contamination would make the results unreliable.
Conclusion
In conclusion, the present report described the clinical application of affected-embryo-based SNP haplotyping with NGS for the PGT of Marfan syndrome, resulting in the birth of a healthy baby. The successful case verified the efficiency of NGS-PGT. It provided an approach for widespread clinical application in monogenetic diseases, especially when there were no offsprings or complete family members for linkage analysis.
Materials and methods
WGA and Sanger sequencing
MDA using a REPLI-g Single Cell Kit (Qiagen, Germany) was performed according to the manufacturer’s protocol. The WGA products were purified and then sequenced. The mutation site of the WGA products was detected using 2× GoldStar Best MasterMix (Dye) (CoWin Biosciences). Forward (5ʹ-GCTTATTTTTCTCCATGGTG-3ʹ) and reverse primer (5ʹ-GTGGAAAAGAGTCAGGATCAT-3ʹ) were designed using Primer 5.0 software. All PCR amplifications were performed on a 96 Well Thermal Cycler Veriti DX (Life Technologies). The amplification reaction was as following: initial denaturation at 94°C for 5 min; 32 cycles of denaturation at 94°C for 1 min, annealing at 62°C for 1 min, extension at 72°C for 2 min. Sanger sequencing was performed on an Applied Biosystems 3500 platform (Life Technologies) after PCR amplification was conducted. All of the procedures were carried out in accordance with the manufacturer’s protocol.
Haplotype construction
One hundred SNP markers located 1 Mb upstream and downstream of the mutation site (chr15:48,736,736 NM_000138.5 (FBN1):c.6037 + 2 T > C) in intron 49 of the FBN1 gene (NM_000138, chr15:48,700,503–48937985) were chosen for NGS-based SNP haplotyping. These SNP sites were submitted to Ion Ampliseq Designer (https://www.ampliseq.com/) for primer design. The genomic DNA (gDNA) of the couple and the WGA products were amplified using these SNP primers to analyze the pedigree. NGS was performed for both SNP haplotyping and mutation locus analysis. Sequencing libraries were prepared using a gene sequencing library kit (NEXTflex Rapid DNA-seq Kit 96rxns, BIOO), and the libraries were sequenced on an Illumina MiseqDX platform (Illumina) using MiSeq Dx Reagent Kit V3 (Illumina). All of the procedures were carried out in accordance with the manufacturer’s protocol. The personal genome machine sequencing data were analyzed by Peking Jabrehoo Med Tech., Ltd. Of the 100 SNPs, 60 informative SNPs including the pathogenic mutation site were selected to establish the haplotype and distinguish the mutant/affected haplotype. Among them, the paternal informative SNP was the site at which the father was heterozygous and the mother was homozygous, while the maternal informative SNP was the site at which the mother was heterozygous and the father was homozygous.
Aneuploidy testing
CNV analysis was performed as previously described (Ou et al. Citation2020). The WGA products were used for library preparation with Easy Prep PGS DNA Lib Prep Kit for Illumina (Peking Jabrehoo Med Tech Co., Ltd). And on the sequencing machine (Miseq, Illumina, Ltd.) according to the instructions of the MiSeq Reagent Kit v3, 150-cycles (Illumina, Inc.). The data from the Miseq sequencing were analyzed by Peking Jabrehoo Med Tech., Ltd. (Beijing, China). In brief, the Illumina MiSeq platform was used for NGS, and approximately 1.5 million fragments of amplified DNA from each TE biopsy were sequenced. After the low-quality bases and adaptors were removed, clean and high-quality reads were compared with the hg19 reference genome. Unique mapped reads were calculated, and a reference dataset was obtained to represent the relative copy number. PGXcloud cloud server (available at http://www.pgxcloud.com/) was used to analyze the chromosomal CNVs (Jabrehoo, China).
Frozen embryo transfer and follow-up assessment
A frozen nonpathogenic euploid blastocyst (E1) grading IVAA was thawed and cultured for 2 h before transfer into the uterus. The embryo grading was assessed following Istanbul consensus (Alpha Scientists in Reproductive M, Embryology ESIGo Citation2011). Clinical pregnancy was defined as the presence of a gestational sac and a fetal heartbeat by sonography at 28 days after FET. Amniocentesis was performed at 19 weeks of gestation to verify the accuracy of the PGT.
Ethics approval
This study was approved by the Reproductive Medical Ethics Committee of Guangzhou Women and Children’s Hospital. Written informed consent was obtained from this couple.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.
Authors’ contributions
Devised the idea for this study: DY.SL; Performed PGT of MFS and wrote the manuscript: OZ, DY; Performed ICSI and the TE biopsy: CZ; Performed clinical work: DY. All authors critically reviewed the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental Material
Download MS Word (25 KB)Acknowledgments
We would like to express our gratitude to the family who participated in the present study. We are extremely grateful to the embryologist and the geneticists of the Guangzhou Women and Children’s Medical Center for contributing to this study.
Supplemental Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Agg B, Benke K, Szilveszter B, Polos M, Daroczi L, Odler B, Nagy ZB, Tarr F, Merkely B, Szabolcs Z. 2014. Possible extracardiac predictors of aortic dissection in Marfan syndrome. BMC Cardiovasc Disord. 14(1):47. doi:10.1186/1471-2261-14-47.
- Alpha Scientists in Reproductive M, Embryology ESIGo. 2011. The Istanbul consensus workshop on embryo assessment: proceedings of an expert meeting. Human Reproduction. 26(6):1270–1283. doi:10.1093/humrep/der037.
- Altarescu G, Brooks B, Kaplan Y, Eldar-Geva T, Margalioth EJ, Levy-Lahad E, Renbaum P. 2006. Single-sperm analysis for haplotype construction of de-novo paternal mutations: application to PGD for neurofibromatosis type 1. Human Reproduction. 21(8):2047–2051. doi:10.1093/humrep/del064.
- Chamayou S, Sicali M, Lombardo D, Alecci C, Ragolia C, Maglia E, Liprino A, Cardea C, Storaci G, Romano S, et al. 2019. Universal strategy for preimplantation genetic testing for cystic fibrosis based on next-generation sequencing. J Assist Reprod Genet. 37(1):213–222. doi:10.1007/s10815-019-01635-2
- Chen L, Diao Z, Xu Z, Zhou J, Yan G, Sun H. 2017. The clinical application of NGS-based SNP haplotyping for PGD of Hb H disease. Syst Biol Reprod Med. 63(3):212–217. doi:10.1080/19396368.2017.1296501.
- Chen L, Diao Z, Xu Z, Zhou J, Yan G, Sun H. 2019. The clinical application of single-sperm-based SNP haplotyping for PGD of osteogenesis imperfecta. Syst Biol Reprod Med. 65(1):75–80. doi:10.1080/19396368.2018.1472315.
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, et al. 1991. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 352(6333):337–339. doi:10.1038/352337a0
- Dietz HC, Pyeritz RE. 1995. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum Mol Genet. 4(suppl_1):1799–1809. Spec No:. doi:10.1093/hmg/4.suppl_1.1799.
- Groth KA, Hove H, Kyhl K, Folkestad L, Gaustadnes M, Vejlstrup N, Stochholm K, Ostergaard JR, Andersen NH, Gravholt CH. 2015. Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet J Rare Dis. 10(1):153. doi:10.1186/s13023-015-0369-8.
- Guo W, Zhu X, Yan L, Qiao J. 2018. The present and future of whole-exome sequencing in studying and treating human reproductive disorders. Journal of Genetics and Genomics = Yi Chuan Xue Bao. 45(10):517–525. doi:10.1016/j.jgg.2018.08.004.
- Handyside AH, Harton GL, Mariani B, Thornhill AR, Affara N, Shaw MA, Griffin DK. 2010. Karyomapping: a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J Med Genet. 47(10):651–658. doi:10.1136/jmg.2009.069971.
- Harper JC, Wilton L, Traeger-Synodinos J, Goossens V, Moutou C, SenGupta SB, Pehlivan Budak T, Renwick P, De Rycke M, Geraedts JP, et al. 2012. The ESHRE PGD Consortium: 10 years of data collection. Hum Reprod Update. 18(3):234–247. doi:10.1093/humupd/dmr052
- Keane MG, Pyeritz RE. 2008. Medical management of Marfan syndrome. Circulation. 117(21):2802–2813. doi:10.1161/CIRCULATIONAHA.107.693523.
- Li H, Qu W, Meng B, Zhang S, Yang T, Huang S, Yuan H. 2012. Identification and study of a FBN1 gene mutation in a Chinese family with ectopia lentis. Mol Vis. 18:504–511.
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, et al. 2010. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 47(7):476–485. doi:10.1136/jmg.2009.072785
- Natesan SA, Bladon AJ, Coskun S, Qubbaj W, Prates R, Munne S, Coonen E, Dreesen JC, Stevens SJ, Paulussen AD, et al. 2014. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 16(11):838–845. eng. doi:10.1038/gim.2014.45.
- Ou Z, Chen Z, Yin M, Deng Y, Liang Y, Wang W, Yao Y, Sun L. 2020. Re-analysis of whole blastocysts after trophectoderm biopsy indicated chromosome aneuploidy. Hum Genomics. 14(1):3. doi:10.1186/s40246-019-0253-z.
- Renwick P, Trussler J, Lashwood A, Braude P, Ogilvie CM. 2010. Preimplantation genetic haplotyping: 127 diagnostic cycles demonstrating a robust, efficient alternative to direct mutation testing on single cells. Reprod Biomed Online. 20(4):470–476. doi:10.1016/j.rbmo.2010.01.006.
- Shen X, Xu Y, Zhong Y, Zhou C, Zeng Y, Zhuang G, Ding C, Li T. 2011. Preimplantation genetic diagnosis for alpha-and beta-double thalassemia. J Assist Reprod Genet. 28(10):957–964. doi:10.1007/s10815-011-9598-5.
- Sueoka K. 2016. Preimplantation genetic diagnosis: an update on current technologies and ethical considerations. Reprod Med Biol. 15(2):69–75. doi:10.1007/s12522-015-0224-6.
- Thornhill AR, Handyside AH, Ottolini C, Natesan SA, Taylor J, Sage K, Harton G, Cliffe K, Affara N, Konstantinidis M, et al. 2015. Karyomapping-a comprehensive means of simultaneous monogenic and cytogenetic PGD: comparison with standard approaches in real time for Marfan syndrome. J Assist Reprod Genet. 32(3):347–356. eng. doi:10.1007/s10815-014-0405-y.
- Wilton L, Thornhill A, Traeger-Synodinos J, Sermon KD, Harper JC. 2009. The causes of misdiagnosis and adverse outcomes in PGD. Human Reproduction. 24(5):1221–1228. doi:10.1093/humrep/den488.
- Xiao Y, Liu X, Guo X, Liu L, Jiang L, Wang Q, Gong B. 2017. A novel FBN1 mutation causes autosomal dominant Marfan syndrome. Mol Med Rep. 16(5):7321–7328. doi:10.3892/mmr.2017.7544.
- Yan L, Wei Y, Huang J, Zhu X, Shi X, Xia X, Yan J, Lu C, Lian Y, Li R, et al. 2014. Advances in preimplantation genetic diagnosis/screening. Sci China Life Sci. 57(7):665–671. doi:10.1007/s11427-014-4683-5
- Yang H, Luo M, Fu Y, Cao Y, Yin K, Li W, Meng C, Ma Y, Zhang J, Fan Y, et al. 2016. Genetic testing of 248 Chinese aortopathy patients using a panel assay. Sci Rep. 6(1):33002. doi:10.1038/srep33002
- Zeevi DA, Zahdeh F, Kling Y, Carmi S, Altarescu G. 2019. Off the street phasing (OTSP): no hassle haplotype phasing for molecular PGD applications. J Assist Reprod Genet. 36(4):727–739. doi:10.1007/s10815-018-1392-1.