
ABSTRACT
This study describes a successful case of preimplantation genetic testing for the monogenic disease (PGT-M) of methylmalonic acidemia (MMA). To avoid the transmission of pathogenic mutations and unnecessary pregnancy termination we applied next-generation sequencing (NGS)-based haplotyping on a couple with a previously deceased MMA offspring. After embryo preparation, all samples were amplified successfully by whole genome amplification. We performed preimplantation genetic testing for aneuploidy (PGT-A) to determine the copy number of embryos’ chromosomes. PGT-A results showed five blastocysts (2, 11, 14, 15 and 16) with balanced chromosomes (46, XN). Two techniques were used for PGT-M. Sanger sequencing was used to detect the mutations of MMUT gene directly, and NGS-based single nucleotide polymorphism (SNP) haplotyping was used to distinguish the chromosomes that carried the mutation. Sanger sequencing and NGS-based SNP haplotyping confirmed that samples 2 and 15 carried c.730insTT, samples 11 and 15 carried c.1105 C > T and samples 14 and 16 did not carry any mutation. Thus, blastocyst 14 was transferred into the mother’s uterus. After prenatal diagnosis at 18 weeks of gestation, a healthy infant without MMUT mutation was born at full term. This study highlights the efficiency of NGS-based SNP haplotyping for PGT-M of MMA.
Abbreviations: MMA: methylmalonic acidemia; MMUT: methylmalonyl-CoA mutase; PGT-M: preimplantation genetic testing for monogenic disease; PGD: preimplantation genetic diagnosis; IVF: in vitro fertilization; ADO: allele dropout; WGA: whole genome amplification; SNP: single nucleotide polymorphism; NGS: next-generation sequencing; PND: prenatal diagnosis; ICSI: intracytoplasmic sperm injection; TE: trophectoderm; DOP-PCR: degenerate oligonucleotide primed polymerase chain reaction; PGT-A: preimplantation genetic testing for aneuploidy; PCR: polymerase chain reaction.
Introduction
Methylmalonic acidemia (MMA) is a group of autosomal recessive disorders mainly divided into isolated MMA and combined MMA with homocysteinemia (Almási et al. Citation2019). The methylmalonyl-CoA mutase (MMUT) type MMA is one of the isolated forms caused by a complete or partial deficiency of MMUT due to mutations in the MMUT (NM_000255.4) gene (Ji et al. Citation2019a). MMUT deficiency causes elevated levels of methylmalonyl-CoA and methylmalonic acid in body fluids and tissues. Delayed diagnosis and treatment results in the accumulation of methylmalonic acid, which damages the central nervous system and causes deep coma, metabolic crisis and death during the newborn period (Tanacan et al. Citation2019; Han et al. Citation2020). Patients with MMA, although treated in accordance with guidelines, including dietary protein restriction, carnitine supplementation and the use of drugs to modulate ammonia, may still experience acute metabolic crisis or even have to undergo surgery (Baumgartner et al. Citation2014; Fraser and Venditti Citation2016). Furthermore, these treatments can neither block the vertical heredity of pathogenic mutations nor guarantee quality of life.
Preimplantation genetic testing for monogenic diseases (PGT-M), which used to be called preimplantation genetic diagnosis (PGD), is a well-established technique for preventing the birth of a child with genetic disorder. PGT-M is performed as part of the in vitro fertilization (IVF) process and used to select unaffected embryos from patients or carriers suffering from known monogenic diseases, such as autosomal recessive, autosomal dominant, and X-linked disorders (Priner et al. Citation2019). The gold standard method for detecting the pathogenic gene mutation is Sanger sequencing. However, allele dropout (ADO) may occur during whole genome amplification (WGA) of PGT-M and may lead to Sanger sequencing failure. To identify ADO and avoid misdiagnosis, we can perform a haplotyping analysis of single nucleotide polymorphism (SNP) markers flanking the mutation site through next-generation sequencing (NGS) (Renwick et al. Citation2006; Hao et al. Citation2018; Masset et al. Citation2019).
This study describes a successful case of applying NGS-based haplotyping in PGT-M to avoid MMA caused by pathogenic mutations of MMUT. We utilized two molecular techniques to ensure the accuracy of PGT-M and help a couple carrying heterozygous mutations of MMUT give birth to a healthy infant.
Results and discussion
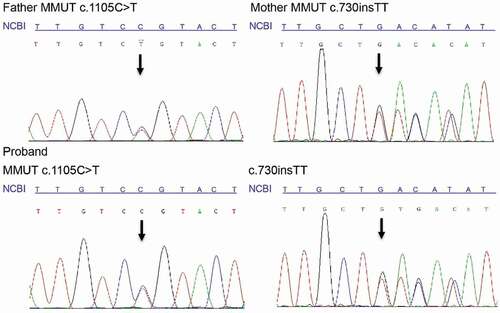
The proband was clinically diagnosed as MMA with markedly elevated propionyl carnitine and ratio of propionyl carnitine/acetyl carnitine through newborn screening. Molecular diagnosis was conducted by Sanger sequencing. The results showed that the father had a heterozygous mutation c.1105 C > T in MMUT, the mother had another heterozygous mutation c.730insTT in MMUT, and the proband inherited both c.1105 C > T and c.730insTT (). Based on the ACMG guidelines, the mutation c.1105 C > T was evaluated as ‘Pathogenic’ (PS3 + PM2 + PM3 + PM5 + PP3 + PP4) and the mutation c.730insTT was also evaluated as ‘Pathogenic’ (PVS1 + PM2 + PP4). After genetic counseling, the couple requested to receive IVF + PGT-M.
Figure 1. Molecular diagnosis results of the family. The loci of the mutations c.730insTT and c.1105 C > T are the nucleotides with a black arrow. Compared with the reference sequence of National Center for Biotechnology Information (NCBI), the father had a heterozygous mutation c.1105 C > T in MMUT, the mother had another heterozygous mutation c.730insTT in MMUT, and the proband inherited both c.1105 C > T and c.730insTT.
Eight blastocysts (2, 4, 11, 13, 14, 15, 16 and 17) were obtained after embryo preparation, and the biopsied cells from these blastocysts were amplified successfully (). We performed preimplantation genetic testing for aneuploidy (PGT-A) on the WGA products. The results indicated that sample 4 had a mosaic deletion on chromosome 6, sample 13 had a mosaic deletion on chromosome 2 (q21.2–q32.1) (56.34 Mb) and sample 17 had a deletion on chromosome 7 (p21.3–q11.23) (67.88 Mb) and a duplication on chromosome 7 (q22.1–q36.1) (49.99 Mb). The rest of the samples’ (2, 11, 14, 15 and 16) chromosomes were balanced (46, XN) ().
Figure 2. Quality control of whole genome amplification (WGA). 2, 4, 11, 13, 14, 15, 16, 17, NC and PC represent sample 2, sample 4, sample 11, sample 13, sample 14, sample 15, sample 16, sample 17, negative control and positive control (PC), respectively. All samples were same with PC that means amplified successfully.

Figure 3. Preimplantation genetic testing for aneuploidy (PGT-A) results of all samples (2, 4, 11, 13, 14, 15, 16 and 17). Compared with the hg19 reference genome through PGT-A analysis, the sketch maps shown that sample 4 has a mosaic deletion of the chromosome 6; Sample 13 has a mosaic deletion in the chromosome 2 (q21.2–q32.1) (56.34 Mb); Sample 17 has a deletion in the chromosome 7 (p21.3–q11.23) (67.88 Mb) and a duplication in the chromosome 7 (q22.1–q36.1) (49.99 Mb). The deletions were marked with red arrows and the duplication was marked with blue arrow. Sample 2’s, 11’s, 14’s, 15’s and 16’s chromosomes were balanced.

Sanger sequencing and SNP haplotyping were used to perform PGT-M on the remaining five samples. The results of Sanger sequencing showed that samples 2 and 15 carried c.730insTT, whereas samples 11, 14 and 16 did not (). However, the sequencing of site c.1105 failed in all the samples possibly because ADO occurred in WGA. The strategy of constructing the pedigree haplotypes is selecting heterozygote SNPs as close to the mutation as possible in the proband. Then SNPs for the couple at the same site were selected to construct the pedigree haplotypes. According to this strategy, we selected 12 informative SNPs from 70 SNPs by analyzing the haplotypes of the proband and her parents in the pre-examination of haplotype analysis (). Then, the haplotypes of the samples (2, 11, 14, 15 and 16), in combination with the pedigree haplotypes, were analyzed. The results showed that samples 2 and 15 carried c.730insTT and samples 11 and 15 carried c.1105 C > T. However, samples 14 and 16 did not carry any MMUT mutation (). Blastocyst 14 was implanted into the mother’s uterus due to its high blastocyst score, and blastocyst 16 was stored in liquid nitrogen for later use.
Table 1. NGS-based SNP haplotyping results of sample 2, 11, 14, 15 and 16
Figure 4. Sanger sequencing results of preimplantation genetic testing for monogenic disease and prenatal diagnosis. (A) Sanger sequencing results of samples (2, 11, 14, 15 and 16). The loci of the mutation c.730insTT are the nucleotides with a black arrow. Compared with the reference sequence of National Center of Biotechnology Information (NCBI) sample 2 and 15 carried the MMUT mutation c.730insTT, while sample 11, 14 and 16 did not. (B) Prenatal diagnosis results of the fetus (from sample 14) by Sanger sequencing. The loci of the mutations c.730insTT and c.1105 C > T are the nucleotides with a black arrow. Compared with the reference sequences of NCBI the fetus neither carried the MMUT mutation c.730insTT nor c.1105 C > T. These results are same to the PGT-M results of sample 14.
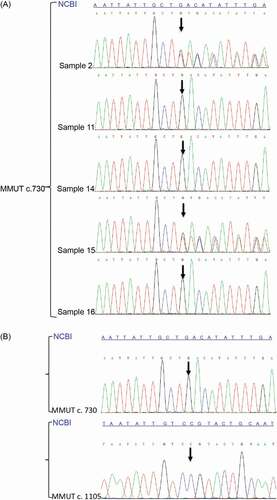
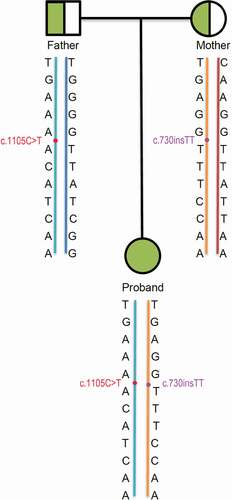
Figure 5. Pedigree of haplotype construction of the MMA family. Circles and square represent females and male, respectively. The half green symbols represent carriers who carried a MMUT mutation and the green symbol represent proband who suffered MMA. The baby blue lines were carrying c.1105 C > T haplotypes, yellow lines were carrying c.730insTT haplotypes, navy blue line and brown line were normal haplotypes. The sites of mutation c.1105 C > T were marked with red points and the sites of mutation c.730insTT were marked with purple points. The carrying c.1105 C > T haplotype of the proband was inherited from her father, and the carrying c.730insTT haplotype was inherited from her mother.
Clinical pregnancy was confirmed through ultrasound after single blastocyst transplantation. Prenatal diagnosis (PND) was conducted by amniocentesis at 18 weeks of gestation. The Sanger sequencing results confirmed that the fetus carried a wild type with sites c.730 and c.1105 (). These results are consistent with those of PGT-M. A genetically healthy infant was born at 40 weeks of gestation.
MMUT-type MMA, which is characterized by MMUT deficiencies in the breakdown pathways of amino acids (isoleucine, valine, methionine, threonine, thymine and odd-carbon-number fatty acids), lead to elevated levels of methylmalonyl-CoA and methylmalonic acid in the organism and result in recurrent vomiting, hypotonia, respiratory distress, multiorgan failure and life-threatening metabolic ketoacidosis. Even with rapid diagnosis and appropriate treatment, the long‐term outcomes of MMA remain disappointing because of neurological and renal complications (Chandler et al. Citation2009; Almási et al. Citation2019; Ji et al. Citation2019a). Furthermore, liver transplantation is regarded as a therapeutic modality, but patients still tend to be metabolically fragile and require metabolism-correcting medications after surgery (Jiang et al. Citation2019).
In the first decade of 2000, the MMA mortality rate was around 40%; thus, MMA is considered an important global health concern, especially in a country as populous as China (Imataka et al. Citation2013; Harrington et al. Citation2016). Although China has no definite national incidence of MMA, MMA is still the most common disease of organic acid metabolism in China; the estimated incidence of MMA is reported to range from 1:26,000 in Beijing and Shanghai to 1:64,708 in Zhejiang Province (Huang et al. Citation2011; Tu et al. Citation2013). In addition, due to the implementation of the two-child policy in 2015 and the three-child policy in 2021 in China, increased number of MMA births expected. Thus, the vital role of genetic diagnosis should be given considerable attention because it can help us identify pathogenic mutations and prevent vertical transmission in carriers of MMA.
PGT-M has been proven to be an efficient approach to help patients with monogenic disorders have healthy offspring. In this study, we reported our experience of applying NGS-based haplotyping in PGT-M to determine MMUT-type MMA. Habibzadeh et al. (Citation2020) reported a similar case for PGT-M of MMA (Habibzadeh et al. Citation2020). However, they only applied Sanger sequencing in PGT-M to determine the WGA products of blastocysts. Although Sanger sequencing is considered the most direct way to detect a point mutation, ADO may still occur in the polymerase chain reaction (PCR) procedure, especially in WGA, and may lead to sequencing failure in WGA products. ADO occurs because one of the two alleles is preferentially amplified, and the other allele is amplified at an undetectable level; thus, sequencing would fail in this condition (Chen et al. Citation2015). In our study, sequencing of site c.1105 failed in all the WGA products, but we used SNP haplotyping to distinguish the mutation-carrying chromosomes. We would not have been able to determine the genotype of c.1105 if we used Sanger sequencing only. Thus, our study highlights the efficiency of using NGS-based SNP haplotyping for PGT-M to avoid the inheritance of MMA caused by MMUT mutations. We try to use two approaches to provide a reference and foundation for future standards, which will guide genetic counseling and the choice of treatment strategy.
Materials and methods
Patient and relatives
A 36-year-old woman (Gestation 3, Parity 1, Abortion 1, Died 1; G3P1A1D1)), who wanted to have a healthy baby with her 37-year-old husband, was referred to our Assisted Reproductive Technology Center for diagnosis. The couple are nonconsanguineous, and their chromosomal karyotypes were normal. They had a girl diagnosed with MMA caused by MMUT mutations in 2013. A compound heterozygous mutation was diagnosed in the MMUT gene with co-segregation. One mutation [c.730insTT (p.Asp244fs)] was inherited from the mother, and another [c.1105 C > T (p.Arg369Cys)] was from the father. After genetic counseling, the couple requested to receive IVF + PGT-M + PND and was advised contraception. Full informed consent was obtained from the couple. This project was approved by the Ethics Committee of Northwest Women’s and Children’s Hospital.
DNA preparation and WGA
Peripheral blood of the proband and her parents was collected in vacuum anticoagulant tubes. DNA was extracted with the RelaxGene Blood DNA System (Tiangen) in accordance with the manufacturer’s standard protocol. The DNA samples were used for genetic diagnosis and to build the haplotyping pedigree.
The embryo preparation included ICSI, embryo culture, TE biopsy of blastocysts and vitrification of post-biopsied blastocysts performed in accordance with the conventional procedures of our embryo laboratory (Ji et al. Citation2019b). The biopsied cells were transferred into 0.2 ml PCR tubes with 4.5 µl of buffer (Peking Jabrehoo Med Tech Co.) for WGA. The degenerate oligonucleotide primed PCR (DOP-PCR) method was applied using the PicoPLEX WGA kit (Rubicon Genomics) to conduct WGA of the biopsy samples in accordance with the manufacturer’s protocol. After assessment by agarose gel electrophoresis, the WGA products were used for PGT-A and PGT-M (including Sanger sequencing and NGS-based SNP haplotyping).
PGT-A
Sequencing libraries were conducted using the gene sequencing library kit (Peking Jabrehoo Med Tech Co.). These libraries were sequenced on the Illumina Miseq platform (Illumina) by using the MiseqDx Universal Kit V3 (Illumina). All procedures were implemented in accordance with the manufacturer’s protocol. After purification and quantification, clean and high-quality reads were analyzed as described previously (Li and Durbin Citation2010). After analyzing the sequencing reads, we reported the deletions/duplications of fragments that were more than 10 Mbp aligned to the reference genome.
Sanger sequencing for PGT-M and PND
Primer 5.0 software was used to design primers of MMUT mutations (c. 730insTT and c.1105 C > T). The primers were as follows: c.730 forward primer (AGCATGTTGTAAAAATTCCTACATTCAAGG) and reverse primer (GAGCAGTTATTCCAGTTCTTGCAAAT). c.1105 forward primer (CAAGGATCAGCCACTTTGGGAA) and reverse primer (TGCAATATCTATCACCTGTTTCTTTGAGTT). PCR and Sanger sequencing were performed as described previously (Ji et al. Citation2019b).
SNP haplotyping for PGT-M
SNP haplotyping was conducted by selecting 70 SNPs within 1 Mbp regions flanking the mutation site. After analyzing the SNPs of the proband and her parents, we selected 12 informative SNPs to establish the pedigree haplotypes. The WGA products were tested in the same manner. By combining the haplotypes of each sample and pedigree haplotypes, we could distinguish the chromosomes carrying the mutations. The purified WGA products were utilized for the construction of sequencing libraries by using the PGD DNA Library Kit for Illumina (Peking Jabrehoo Med Tech Co.). Then, the purified and quantified libraries were sequenced on the Illumina Miseq platform (Illumina). All procedures were performed in accordance with the manufacturer’s protocols. The NGS sequencing data were analyzed by Peking Jabrehoo Med Tech Co., Ltd.
Ethics approval
This study was approved by the Ethics Committee of Northwest Women and Children’s Hospital (No. 2020–016). All patients provided written informed consent.
Authors’ contributions
Conceived and designed the experiments: SZ, RQ; performed the experiments: BH, QWh; acquisition the data: XJ, WS, JS; analyzed the data: BH; wrote the manuscript: BH, LW.
Acknowledgments
Firstly, we would like to express our gratitude to the family for participating in this study; Changsheng Wu of Peking Jabrehoo Med Tech Co., Ltd., for his work; and the embryologist and the geneticists of the Northwest Women’s and Childre’s Hospital for contributing to this study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Almási T, Guey LT, Lukacs C, Csetneki K, Vokó Z, Zelei T. 2019. Systematic literature review and meta-analysis on the epidemiology of methylmalonic acidemia (MMA) with a focus on MMA caused by methylmalonyl-CoA mutase (mut) deficiency. Orphanet J Rare Dis. 14(1):84. doi:https://doi.org/10.1186/s13023-019-1063-z.
- Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, Huemer M, Hochuli M, Assoun M, Ballhausen D, et al. 2014. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 9:130.
- Chandler RJ, Zerfas PM, Shanske S, Sloan J, Hoffmann V, DiMauro S, Venditti CP. 2009. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J. 23(4):1252–1261. doi:https://doi.org/10.1096/fj.08-121848.
- Chen YF, Jia HT, Chen ZH, Song JP, Liang Y, Pei JJ, Wu ZJ, Wang J, Qiu YL, Liu G, et al. 2015. Mutational spectrum of phenylketonuria in Jiangsu province. Eur J Pediatr. 174(10):1333–1338. doi:https://doi.org/10.1007/s00431-015-2539-z
- Fraser JL, Venditti CP. 2016. Methylmalonic and propionic acidemias: clinical management update. Curr Opin Pediatr. 28(6):682–693. doi:https://doi.org/10.1097/MOP.0000000000000422.
- Habibzadeh P, Tabatabaei Z, Farazi Fard MA, Jamali L, Hafizi A, Nikuei P, Salarian L, Nasr Esfahani MH, Anvar Z, Faghihi MA. 2020. Pre-implantation genetic diagnosis in an Iranian family with a novel mutation in MUT gene. BMC Med Genet. 21(1):22. doi:https://doi.org/10.1186/s12881-020-0959-8.
- Han B, Nie W, Sun M, Liu Y, Cao Z. 2020. Clinical presentation, molecular analysis and follow-up of patients with mut methylmalonic acidemia in Shandong province, China. Pediatr Neonatol. 61(2):148–154. doi:https://doi.org/10.1016/j.pedneo.2019.07.004.
- Hao Y, Chen D, Zhang Z, Zhou P, Cao Y, Wei Z, Xu X, Chen B, Zou W, Lv M, et al. 2018. Successful preimplantation genetic diagnosis by targeted next-generation sequencing on an ion torrent personal genome machine platform. Oncol Lett. 15(4):4296–4302.
- Harrington EA, Sloan JL, Manoli I, Chandler RJ, Schneider M, McGuire PJ, Calcedo R, Wilson JM, Venditti CP. 2016. Neutralizing Antibodies Against Adeno-Associated Viral Capsids in Patients with mut Methylmalonic Acidemia. Hum Gene Ther. 27(5):345–353. doi:https://doi.org/10.1089/hum.2015.092.
- Huang XW, Yang JB, Tong F, Yang RL, Mao HQ, Zhou XL, Huang XL, Yang LL, Huang CG, Zhao ZY. 2011. [Screening for neonatal inborn errors of metabolism by electrospray ionization-tandem mass spectrometry and follow-up]. Zhonghua Er Ke Za Zhi = Chinese Journal of Pediatrics. 49(10):765–770.
- Imataka G, Sakamoto O, Yamanouchi H, Yoshihara S, Omura-Hasegawa Y, Tajima G, Arisaka O. 2013. Novel c.2216T > C (p.I739T) mutation in exon 13 and c.1481T > A (p.L494X) mutation in exon 8 of MUT gene in a female with methylmalonic acidemia. Cell Biochem Biophys. 67(1):185–187. doi:https://doi.org/10.1007/s12013-013-9532-9.
- Ji X, Wang H, Ye J, Qiu W, Zhang H, Liang L, Xiao B, Dai M, Xu Y, Chen T, et al. 2019a. Prenatal diagnosis of methylmalonic aciduria from amniotic fluid using genetic and biochemical approaches. Prenat Diagn. 39(11):993–997. doi:https://doi.org/10.1002/pd.5519
- Ji X, Zhang Z, Shi J, He B. 2019b. Clinical application of NGS-based SNP haplotyping for the preimplantation genetic diagnosis of primary open angle glaucoma. Syst Biol Reprod Med. 65(3):258–263. doi:https://doi.org/10.1080/19396368.2019.1590479.
- Jiang YZ, Sun LY, Zhu ZJ, Wei L, Qu W, Zeng ZG, Liu Y, Tan YL, He EH, Xu RF, et al. 2019. Perioperative characteristics and management of liver transplantation for isolated methylmalonic acidemia-the largest experience in China. Hepatobiliary Surg Nutr. 8(5):470–479. doi:https://doi.org/10.21037/hbsn.2019.03.04
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 26(5):589–595. doi:https://doi.org/10.1093/bioinformatics/btp698.
- Masset H, Zamani Esteki M, Dimitriadou E, Dreesen J, Debrock S, Derhaag J, Derks K, Destouni A, Drüsedau M, Meekels J, et al. 2019. Multi-centre evaluation of a comprehensive preimplantation genetic test through haplotyping-by-sequencing. Human Reprod. 34(8):1608–1619. doi:https://doi.org/10.1093/humrep/dez106
- Priner S, Altarescu G, Schonberger O, Holzer H, Rubinstein E, Dekel N, Peretz A, Eldar-Geva T. 2019. The effect of repeated biopsy on pre-implantation genetic testing for monogenic diseases (PGT-M) treatment outcome. J Assist Reprod Genet. 36(1):159–164. doi:https://doi.org/10.1007/s10815-018-1359-2.
- Renwick PJ, Trussler J, Ostad-Saffari E, Fassihi H, Black C, Braude P, Ogilvie CM, Abbs S. 2006. Proof of principle and first cases using preimplantation genetic haplotyping–a paradigm shift for embryo diagnosis. Reprod Biomed Online. 13(1):110–119. doi:https://doi.org/10.1016/S1472-6483(10)62024-X.
- Tanacan A, Gurbuz BB, Aydin E, Erden M, Coskun T, Beksac MS. 2019. Prenatal Diagnosis of Organic Acidemias at a Tertiary Center. Balkan J Med Genet. 22(1):29–34. doi:https://doi.org/10.2478/bjmg-2019-0003.
- Tu WJ, Chen H, He J. 2013. Methylmalonic aciduria: newborn screening in mainland China? J Pediatr Endocrinol Metab. 26(3–4):399–400. doi:https://doi.org/10.1515/jpem-2012-0276.