
Abstract
PIWI-interacting RNAs (piRNAs) are 24–32 nucleotide RNA sequences primarily expressed in germ cells and developing embryos that suppress transposable element expression to protect genomic integrity during epigenetic reprogramming events. We characterized the expression of piRNA sequences and their encoding clusters in sperm samples from an idiopathic fertility model of Holstein bulls with high and low Sire Conception Rates. The piRNA populations were determined to be mostly similar between fertility conditions when investigated by principal component and differential expression analysis, suggesting that a high degree of conservation in the piRNA system is likely necessary for the production of viable sperm. Both fertility conditions demonstrated evidence of ‘ping-pong’ activity – a secondary biogenesis pathway associated with active transposable element targeting and suppression. Most sperm-borne piRNAs were between 29-30 nucleotides in length and originated from 226 clusters across the genome, with the exception of chromosome 20. Mapping analysis revealed abundant targeting of several transposable element families, suggesting a suppressive function of sperm piRNAs consistent with their established roles. Expression of genes targeted by sperm-borne piRNAs is significantly reduced throughout early embryogenesis compared to the mRNA population. Limited transposable element expression is known to be essential for spermatogenesis, thus epigenetic regulation of this pathway is likely to influence sperm quality and fertilizing capacity.
Introduction
Initially identified in Drosophila (Lin and Spradling Citation1997), piRNAs are 24–32 nucleotide sequences most widely expressed in germ cells and developing embryos. They associate with PIWI proteins, classically to target RNA interference mechanisms aimed at the suppression of transposable elements (TEs) (Carmell et al. Citation2007; Roovers et al. Citation2015; Russell et al. Citation2017), but are also capable of targeting mRNAs (Russell et al. Citation2017). TEs are mobile genetic sequences (‘jumping genes’) in DNA that can autonomously replicate and re-insert into the genome, with the potential to cause genetic damage. TEs are normally epigenetically silenced, but often become expressed when methylation and other epigenetic marks are removed in the course of reprogramming events during gametogenesis and embryogenesis (Morgan et al. Citation2005; Bui et al. Citation2009). In these contexts, the PIWI pathway is thought to constrain the expression of most TEs to preserve genomic integrity (Carmell et al. Citation2007; Roovers et al. Citation2015), though it should be noted that some expression of TEs appears essential for successful embryonic development and gamete development (Senft and Macfarlan Citation2021).
During spermatogenesis primary piRNA biogenesis occurs in two distinct phases. First, ‘pre-pachytene piRNAs’ are transcribed from repetitive DNA loci (Aravin et al. Citation2007; Aravin et al. Citation2008; Fu and Wang Citation2014; Han and Zamore Citation2014)—most of which are derived from expressed transposable elements and mRNA sequences (Aravin et al. Citation2008; Fu and Wang Citation2014). The second phase occurs after the completion of pachytene prophase I. These ‘pachytene piRNAs’ are produced from piRNA clusters: repetitive genomic regions densely encoded with piRNAs, transcribed as long primary piRNAs (pri-piRNAs) (Brennecke et al. Citation2007; Han and Zamore Citation2014). Pri-piRNAs are processed to mature piRNAs, which associate with Argonaute proteins of the PIWI clade to form piRNA-induced silencing complexes (piRISC). PiRISC silences gene expression by targeting complementary sequences in transposable elements and other RNA transcripts including mRNAs (Gou et al. Citation2014; Pantano et al. Citation2015; Russell et al. Citation2017), which are subsequently cleaved by endonuclease activity of the PIWI protein (Liu et al. Citation2004).
piRNAs can also be generated by a secondary biogenesis pathway known as the ping-pong amplification loop. In this pathway PIWI proteins cleave RNA targets and use the cleaved product as a template to generate additional ‘secondary’ piRNAs in sense orientation. These sense piRNAs in turn proceed to direct cleavage of antisense targets, producing antisense piRNAs which feed forward into the degradation process (Kelleher and Barbash Citation2013). Targets are cleaved with 10 nucleotide overlaps from the original sequence, increasing piRNA abundance and facilitating expedited suppression of abundant RNA targets. This process produces a distinct ping-pong signature that can be bioinformatically detected by interrogating the frequency of 10 nucleotide overlaps in complementary sequences, with a bias for 5’ uracil and an adenine residue in the 10th position (a consequence of the 10 nucleotide overlap) (Brennecke et al. Citation2007; Han and Zamore Citation2014; Czech and Hannon Citation2016; Russell et al. Citation2017).
piRNAs have been employed as biomarkers to predict different semen quality characteristics. Levels of specific piRNAs in human seminal plasma have been associated with azoospermia (Hong et al. Citation2016), while others within spermatozoa themselves are correlated with sperm concentration and fertilization potential (Cui et al. Citation2018). It is not yet known whether these differences reflect a residual legacy of PIWI/piRNA and TE interactions during spermatogenesis or whether the sequences have functional activity when entering the oocyte at fertilization, although both possibilities are recognized.
In livestock industries, careful reproductive management is essential to maintain efficient production. Artificial insemination is a common practice in the dairy industry, where a low number of genetically superior bulls service a large population of females. To optimize fertility, bulls are selected for high sire conception rate (SCR), and semen samples are subjected to rigorous testing to ensure quality. Currently, Computer Assisted Semen Analysis is the gold standard for quality analysis, however, this technology can predict less than 60% of the variation in fertility (Saacke Citation2008; Utt Citation2016), and some bulls which pass stringent analysis standards may perform poorly in the field. Several other quality assessments are available, including assessments of membrane integrity, mitochondrial activity, and DNA fragmentation (Alves et al. Citation2020), however, limitations are present in all of these approaches. Recent work has examined the expression of small non-coding RNAs in bovine semen (Sellem et al. Citation2020), and our group has previously identified differentially correlated pairs of microRNAs (miRNAs) associated with bull fertility (Werry et al. Citation2022). In the present study, we examined the expression patterns of piRNAs within bovine sperm, wherein associations with fertility may reflect their influence on spermatogenesis and/or early embryonic development.
Results
Read characteristics
Bioinformatic analysis of high throughput RNA sequencing of sperm samples from high (SCR: 2.1 ± 0.7, n = 7) and low (SCR: -2.7 ± 1.1, n = 6) fertility bulls using a modification of our previously-established pipeline (Russell et al. Citation2017) revealed piRNAs to be the 2nd most abundant class of sncRNA, as noted in our previous work investigating the miRNA profile with this dataset (Werry et al. Citation2022). 23,643,300 putative piRNA reads ranging from 24-32 nucleotides in length were present in the samples, with the majority of these reads 29-30 nucleotides long. The distribution of read lengths was independent of fertility conditions in our bull populations ().
Figure 1. Read length distribution and piRNA cluster expression in bovine sperm samples. A comparison of piRNA characteristics between high (red) and low (blue) fertility bulls. (A) Relative abundance of piRNA read lengths within the standard size of piRNAs: 24 – 32 nucleotides. (B) Number of reads mapping to documented piRNA clusters on each chromosome in high and low fertility bulls.
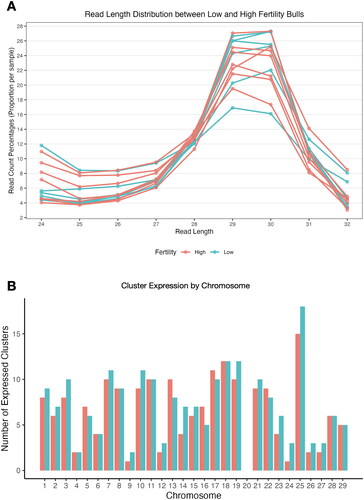
Bovine piRNAs are encoded in 425 clusters in the genome (Rosenkranz et al. Citation2022). We observed piRNAs from 226 of these clusters in sperm. piRNAs from 19 specific piRNA clusters were expressed exclusively in high fertility bulls while piRNAs from 37 different clusters were expressed exclusively in low fertility bulls, though none of these fertility-exclusive sequences were expressed by all bulls in the given condition. The remaining 170 clusters were common to both groups (Table S1). No piRNAs derived from clusters on chromosome 20 were observed in either fertility condition, while the chromosome with the highest number of expressed piRNAs was chromosome 25 ().
Ping-Pong amplification
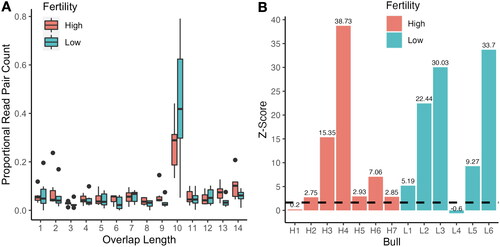
Putative piRNA sequencing reads were investigated for the presence of a ‘ping-pong’ signature, indicative of secondary piRNA biogenesis. Ping-pong amplification is bioinformatically reflected by a distinct 10 nucleotide ‘overlap’ peak between 5’ ends of a subset of sequenced piRNAs. Both high and low fertility animals displayed a high proportion of 10 nucleotide overlaps between reads, suggesting that ping-pong amplification is present and generally similar in both fertility groups. No significant differences (p = 0.19) were observed in the ping-pong signatures between fertility conditions (). Interestingly, while the majority of sperm from both high and low fertility bulls displayed ping-pong activity, Z-scores comparing the frequency of 10 nt overlaps to other overlap lengths indicate that one bull in each condition did not appear to have significant secondary piRNA production (), implying that ping-pong activity is variable and is not directly associated with fertility in all cases in our model. Furthermore, we observed a first-position uracil bias, but not a 10th position adenine bias that is commonly associated with ping-pong activity (Figure S1)
Figure 2. Ping-pong signature comparison between high and low fertility bulls. A comparison of ping-pong signature features between high (red) and low (blue) fertility bulls. (A) Box and whisker plots depicting the proportion of read pairs with specified overlap length between 5’ ends ranging from 1-14. (B) Z-score of 10 nt 5’ overlapping piRNAs for each bull, dashed line indicating significance threshold of Z-score ≥ 1.65 corresponding to one tailed p < 0.05.
piRNA biogenesis through the ping-pong pathway may induce an additional form of primary processing of targeted transcripts by the nuclease PLD6 (known as Zucchini in Drosophila), leading to the production of ‘phased’ piRNAs from consecutive nuclease slicing activity at each base downstream from the ping-pong target site. We did not observe classical hallmarks of piRNA phasing (i.e. a peak of 3’-5’ sequences with a mapped difference of 1 nt, with broader peaks at ∼-32 to ∼-24 nt). Our results do not show a peak at 1, but do reveal a broad double peak at -30/-31 and -27/-26, suggesting that many fragments with identical 5’ ends and varying sequence length are present (Figure S2).
Differential expression of piRNAs
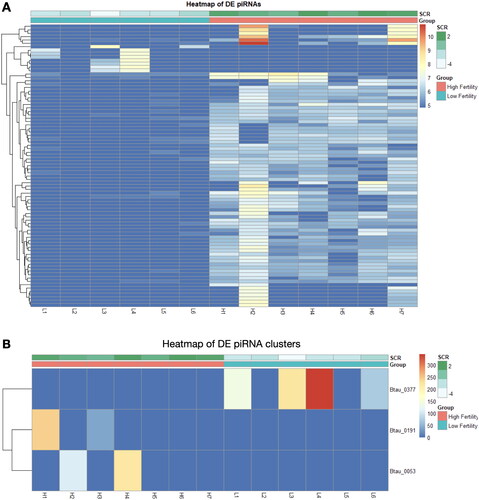
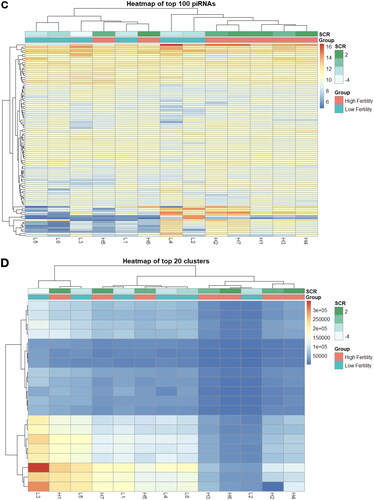
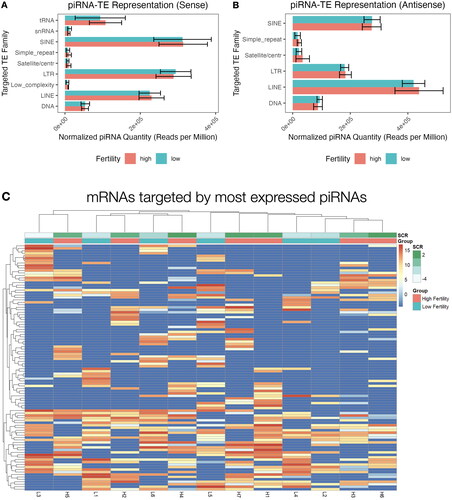
Overall patterns of piRNA expression were compared using PCA, revealing no broad differences in expression of piRNA clusters (Figure S3A) or individual sequences (Figure S3B) between fertility conditions. Differential expression (DE) analysis of fertility groups was also performed at the individual sequence and cluster levels, to identify differences in expression of specific piRNAs. Several DE piRNAs were identified, the majority of which displayed elevated expression in the high fertility group compared to low fertility (). Due to a lack of consistency in piRNA nomenclature, DE piRNA sequences and names available from piRBase are also provided in Table S2. Three DE piRNA clusters were identified, however these clusters exhibited low expression and were only present in the sperm of a limited number of bulls from each fertility group (). The majority of the DE piRNAs were not expressed at all in the opposite group, which was also the case for piRNA clusters. Notably, there appears to be substantial variation in the distribution of these DE piRNAs within a specific fertility group, particularly in expression of specific piRNAs in bull H2. Reanalysis including/excluding bull H2 resulted in similar differential expression results. Thus, it appears that a lack of heterogeneity in cluster expression leaves H2 as an apparent outlier, however, it is sufficiently homogenous with the other samples to remain in the sample group. This homogeneity is apparent when using hierarchical clustering to investigate both individual sequences () and clusters (), which revealed no association by fertility status. Expression of the 100 most abundant piRNAs in bovine sperm is available in Table S3.
Figure 3. piRNA expression heatmaps of differentially expressed and highly abundant piRNA sequences and clusters. Differentially expressed piRNAs between high (red) and low (blue) fertility animals when analyzed by sequences (A) and clusters (B). Hierarchical clustering of the 100 highest expressed piRNA sequences (C) and 20 highest expressed clusters (D), as shown below.
Predicted targets of sperm piRNAs
To further explore their potential functional roles in sperm and spermatogenesis, piRNAs were mapped to specific transposable elements annotated using RepeatMasker. LINE, SINE, and LTR elements were most commonly targeted by piRNAs in sperm and targeting in both sense and antisense orientations was observed (). No individual transposable element families appeared differentially targeted by piRNAs in sperm from either fertility condition. Interestingly, there is substantial heterogeneity with respect to which specific members of each TE family are targeted across bulls in both groups, and hierarchical clustering revealed no significant specific trends between the fertility groups ().
Figure 4. Mapped transposable element targets of expressed piRNAs. Reads mapped to repetitive elements (tRNA: transfer RNA; snRNA: small nuclear RNA; SINE: short interspersed nuclear element; LTR: long terminal repeat; LINE: long intersperced nuclear element) in sense (A) and antisense (B) orientation, read counts are ± SD. (C) Hierarchical clustering the top 100 most targeted repetitive elements by expression of mapped piRNAs in each sample.
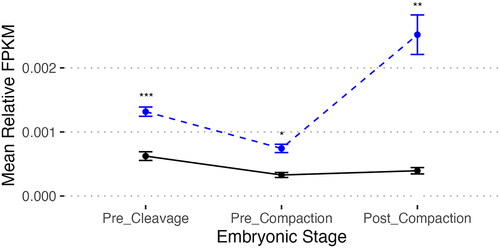
In addition to their classical roles targeting TEs, bovine piRNAs have also been observed to target mRNAs (Russell et al. Citation2017). In our samples, we investigated piRNAs mapping to somatic genes in three contexts. Sequence-specific expression data including mRNA mapping of individual targets is available in Table S4. First we mapped piRNAs to the 3‘UTRs (the typical site of RNA interference mapping for microRNAs and other small RNA sequences), identifying 1356 mapped genes. We also mapped to spliced exons of post-processed mRNA, revealing 1582 genes, and the unspliced mRNA transcript sequence, revealing 2325 genes (Table S5). No significant differential targeting of mRNAs between fertility conditions was observed. The bovine embryoGENE Profiler was employed to investigate the expression of these mRNA transcripts during early embryogenesis (Robert et al. Citation2011; Khan et al. Citation2016). This analysis revealed that piRNAs expressed in all bulls studied map to mRNAs that are significantly more abundant in pre-cleavage embryos compared to later developmental stages (pre-compaction and post-compaction), while all targeted genes are less expressed than the mean of all genes ().
Figure 5. Relative expression of mRNA transcripts targeted by piRNAs in all samples. Mean relative fragments per kilobase million of mRNA expression at key stages in embryonic development for mRNA transcripts with piRNA targeting predicted by bedtools. Bonferroni-corrected two-tailed pairwise T-test of piRNA-mapped mRNAs (solid black line) in pre-cleavage embryos (GV, MII, 1-cell) are more abundant than those in pre-compaction embryos (2-cells, 4-cells, 8-cells-early, 8-cells-late; p = 1.7e-11) and post-compaction embryos (morula, blastocyst-early, blastocyst late; p = 3.5e-7). Baseline expression of all genes (blue dashed line) indicates an increase in expression in Post_Compaction embryos from Pre_Cleavage (p = 1.6e-5) and Pre_Compaction (p = 5.4e-11). T-test indicates that targeted genes are significantly less expressed than baseline at all timepoints (Pre_Cleavage p = 6.59e-12 (***); Pre_Compaction p = 4.312e-8 (*), Post_Compaction p = 1.188e-11 (**)). mRNA expression data from the EmbryoGENE Profiler (Robert et al. Citation2011; Khan et al. Citation2016).
Discussion
Male fertility relies on complex regulatory pathways that orchestrate spermatogenesis which culminates in the formation of sperm capable of delivering nucleic acids to the oocyte for fertilization (Sharma Citation2019). One pathway that is critically important to developing gametes and embryos is the PIWI/piRNA pathway, which functions to constrain TE expression during reprogramming in addition to other roles including epigenetic gene regulation and direct messenger RNA silencing (Czech and Hannon Citation2016; Russell et al. Citation2017; Ozata et al. Citation2019). piRNAs generated during spermatogenesis may reflect a legacy of cellular events (e.g. TE and target mRNA expression levels) and represent potential biomarkers for fertility. While differences in male fertility with known phenotypes (e.g. azoospermia, epididymitis) have been associated with altered piRNA patterns in seminal fluid and epididymal biopsies (He et al. Citation2022; Gong et al. Citation2022), differences associated with more subtle fertility differences have not been investigated. This is particularly relevant in the context of bovine artificial insemination, as piRNAs are known to direct DNA methylation in developing gametes (Russell et al. Citation2017) and the bovine sperm methylome is strongly associated with fertility in a large population of bulls (Costes et al. Citation2022). In the present study, we sought to determine whether differences in piRNA expression were present in bulls with subtle fertility differences.
The idiopathic differences in fertility present in this model do not appear to be driven by differences in piRNA expression. We postulate that this is a reflection of the essential nature of piRNAs in spermatogenesis and male fertility. All bulls in this study, even those with low fertility, produced morphologically normal sperm with good progressive motility (Werry et al. Citation2022) – characteristics which likely require expression of functional piRNAs. Large-scale changes in piRNA populations in a given bull would therefore be expected to significantly alter measurable parameters of sperm quality, which would lead to their exclusion from the groups studied here. Indeed, we observed several features to be consistent across all bovine sperm samples. piRNA reads (24-32 nucleotides) were most commonly 29-30 nucleotides in length, consistent with another investigation of bovine piRNAs (Sellem et al. Citation2020). Differential expression analysis did identify 83 individual piRNAs and 3 piRNA clusters with significantly different expression patterns between our two groups. While these are statistically significant, we suspect these piRNAs are unlikely to be biologically relevant due to their low overall expression. In many cases the significance appears to be driven by high expression in a limited number of bulls; in the case of the piRNA clusters, one specific cluster was expressed in no more than 4 of the 13 bulls studied. We postulate that, in addition to underlying genetic differences, the variable expression patterns observed are a result of bulls reacting to different environmental changes, emphasized by the variability of targeted repeats. A degree of transcriptomic plasticity is likely essential for piRNAs to appropriately regulate environmentally-dependent changes in TE and mRNA expression during sperm development.
The presence of a ‘ping-pong’ signature in piRNAs present in the sperm samples studied here suggests an active PIWI pathway, as secondary piRNAs are generated after targeted degradation of TE transcripts by the RNAse ‘slicer’ activity of piRISC complexes containing primary piRNAs. This variability may reflect exposure to various environmental stressor or other ‘random’ triggers which drive primary piRNA expression that, in turn, induces a secondary ping-pong response. We suspect that any failure to properly initiate this defence mechanism would lead to significantly decreased fertility (outside the relatively small differences between bulls in this study) in circumstances where TE expression was elevated. Therefore, it is perhaps not surprising that dramatic differences in piRNA expression were not observed in the present study.
The piRNAs identified in the current study demonstrate widespread targeting of LINE, SINE, and LTR TEs, an expected function of piRNAs in developing gametes (Russell et al. Citation2017). This is consistent with our prediction that piRNAs in our model perform indispensable roles in TE control, as regulation of these transposable elements is thought to be the primary mechanism through which piRNAs influence fertility. There is substantial variability in which TE classes are targeted in each bull, likely reflecting the differences in TE expression to which the piRNAs respond. This variability, coupled with the limited differences between the fertility conditions lead us to conclude that differential expression of piRNAs is not a major factor underlying the 4.8 SCR difference observed in these populations.
piRNA expression is critical for successful mammalian spermatogenesis (Sellem et al. Citation2021), and it is likely that significant disruptions would prevent production of viable sperm – a much more pronounced phenotype than the subtle fertility difference investigated here. Whether ping-pong amplification is an essential aspect of this process is unclear – differences in the signature observed in mature sperm may not reflect processes that occur during spermatogenesis. In addition to TE suppression, piRNAs are associated with key developmental events including mRNA turnover during spermiogenesis (Gou et al. Citation2014), instrumental to development of visibly competent sperm. Indeed, previous investigations identifying piRNA differences associated with fertility are associated with recognized fertility phenotypes (Hong et al. Citation2016; He et al. Citation2022). The differences that were observed may reflect variable responses of individual bulls to their environments, which has previously been observed to influence piRNAs in sperm (reviewed in Donkin and Barrès Citation2018).
Interestingly, piRNAs expressed in all samples (and thus considered to be ‘essential’) map to mRNAs expressed highly in pre-compaction embryos. This is similar to our previous findings on the expression of sperm-borne miRNA targeted mRNAs, which had higher expression in all pre-compaction embryos. This more limited period window of abundance for piRNA targets suggests that piRNAs may function to initiate sequence degradation earlier upon delivery to the oocyte. To better understand the roles of piRNAs, it will ultimately be necessary to investigate physiological and pathological induction of both target TE and piRNA expression during spermatogenesis and early embryonic development. Similarly to piRNAs, TEs are known to respond to environmental stressors (Miousse et al. Citation2015) and regulatory interactions between these sequences may present a promising avenue to determine some underlying causes of idiopathic male subfertility in bovine, particularly those involving environmental effects in the course of spermatogenesis.
The high abundance of piRNAs in sperm documented here, coupled with their known roles in mammalian spermatogenesis models (Loubalova et al. Citation2021) provide circumstantial evidence that piRNAs play important roles in the development of viable bovine sperm. Although they do not appear to be major factors in the subtle fertility differences present in the populations studied here, our studies reveal that the expression characteristics and patterns of piRNAs are largely consistent between mature Holstein bulls (mean age in days: high fertility = 967 ± 264; low fertility = 898 ± 102) with acceptable sperm quality as measured by traditional metrics.
Methods
Library preparation and sequencing
Samples were selected for difference in Sire Conception Rate (SCR) – a metric calculated to reflect percentage of fertilizing success compared to a ‘standard’ bull under similar conditions. Processed semen straws from 7 high fertility (HF) bulls with SCR ≥ 1.0, (mean = 2.1 ± 0.7) and 6 low fertility bulls with SCR ≤ –1.0 (mean = –2.7 ± 1.1) were provided by Semex (Guelph, Ontario, Canada). These samples all passed standard quality control analysis and represent a model of idiopathic subfertility. We have previously published an investigation of the miRNA population within these samples in which we provide a detailed description of the methodology (Werry et al. Citation2022). In brief, motile sperm were isolated from 500 µL of cryopreserved semen by Percoll gradient. The motile fraction was snap frozen in liquid nitrogen before storage at –80 °C. NucleoSpin miRNA Extraction Kit (Macherey-Nagel: Düren, Germany) was used to extract total RNA from the frozen sperm pellets. Subsequently, small RNA libraries were generated via NEXTflex Small RNA-Seq Kit v3 (PerkinElmer: Waltham, MA, USA) and size-selected to 140-170 bp by Pippin Prep (Sage Science: Beverly, MA, USA). Libraries were sequenced on Illumina NextSeq high output kit, producing 81,250,738 reads.
Sequence annotation
This study employed a custom BASH pipeline with high performance data processing performed on Digital Research Alliance of Canada’s Graham cluster. A visual workflow of this pipeline is available in Supplementary Figure 1. Data visualization was performed using R, identified scripts were obtained from the NGS toolbox collection (Rosenkranz et al. Citation2015). Unitas was used to annotate FASTQ reads while collapsing and filtering low complexity reads via the Duster algorithm. Unannotated reads (containing piRNAs) were then filtered via the length-filter.pl perl script, discarding reads outside the piRNA range of 24 nt – 32 nt. Read statistics were calculated with the FASTX-toolkit and basic-analyses.pl perl script. Filtered reads were mapped to bovine reference genome bosTau9.fa using sRNAmapper. The output map files contain piRNA coordinates in ELAND format, used to calculate ‘ping-pong’ signature overlap lengths via the pingpong.pl perl script. Map files were also used to calculate the counts of small RNAs derived from repeat elements through input for a custom perl script RMvsMAP.pl which compares mapped piRNA loci with RepeatMasker annotation.
Cluster data were collected by assigning reads by genomic coordinate to clusters identified in the Bos taurus ARS-UCD1.2 genome assembly in the piRNA cluster database (https://www.smallrnagroup.uni-mainz.de/piRNAclusterDB/ (Rosenkranz et al. Citation2022)). Approximately 93% of unique sequences mapped to a cluster. Read counts were calculated as proportions per sample for the read length distribution plot seen in . Read counts were also normalized between 0 and 1 for the ‘ping-pong’ overlap length distribution plot in . Repeat targets with less than 10 mapped hits were filtered out for both and .
Differential expression analysis
Sequences without other small RNA annotations, between 24-32 nt long, and mapping to the genome were considered piRNAs. Raw count matrices were imported into R for differential expression analysis with DESeq2 (Love et al. Citation2014). Sequences with fewer than 5 reads across all samples, or present in fewer than 3 samples were excluded from the analysis. Fertility group was used as the condition in the design formula, and piRNAs were considered differentially expressed below an adjusted p-value of 0.05.
piRNA-mRNA mapping
Mapped piRNAs from the annotation step above were subsequently intersected with coding regions utilizing bedtools intersect with the following settings: -S -wa -wb -f 1 -bed -a piRNA_candidates.bam -b bosTau9.ncbiRefSeq.gtf. Anti-sense intersecting reads falling into gene 3’ UTRs, exons, and across the entire transcript length were compiled into separate matrices for analysis. For each matrix, mean quantity of piRNAs targeting each mRNA were compared by t-test and p-values were Bonferroni corrected to account for multiple tests.
Ethics approval
The research presented here is in full compliance with all relevant codes of experimentation and legislation.
Authors’ contributions
Conceptualization: NW, SR, SM, KH, SL, ML, CL, JL; Methodology: NW, SR, JL; Investigation: NW, SR; Analysis: NW, SR, RS; Resources: SM, KH, SL, ML, CL; Writing – original draft preparation: NW; Writing – review and editing: NW, SR, RS, SM, KH, CL, JL.
| Abbreviations | ||
| PIWI | = | P-element induced wimpy testis |
| RNA | = | Ribonucleic acid |
| piRNA | = | Piwi-interacting RNA |
| mRNA | = | Messenger RNA |
| DNA | = | Deoxyribonucleic acid |
| TE | = | Transposable element |
| Pri-piRNA | = | Primary piRNA |
| SCR | = | Sire conception rate |
| CASA | = | Computer-assisted semen analysis |
| PCA | = | Principal component analysis |
| UTR | = | Untranslated region |
Supplemental Material
Download Zip (10 MB)Disclosure statement
The authors report there are no competing interests to declare. This work was included as part of N Werry’s PhD thesis, available at https://atrium.lib.uoguelph.ca/items/8c377aba-5cab-4614-be8b-3f7f15c45f4b
Data availability statement
The data that support the findings of this study are openly available at BioProject: PRJNA777266.
Additional information
Funding
References
- Alves MBR, Celeghini ECC, Belleannée C. 2020. From sperm motility to sperm-borne microRNA signatures: new approaches to predict male fertility potential. Front Cell Dev Biol. 8:791. doi: 10.3389/fcell.2020.00791.
- Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. 2007. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 316(5825):744–747. doi: 10.1126/science.1142612.
- Aravin AA, Sachidanandam R, Bourc’his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ. 2008. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell. 31(6):785–799. doi: 10.1016/j.molcel.2008.09.003.
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in drosophila. Cell. 128(6):1089–1103. doi: 10.1016/j.cell.2007.01.043.
- Bui LC, Evsikov AV, Khan DR, Archilla C, Peynot N, Hénaut A, Le Bourhis D, Vignon X, Renard JP, Duranthon V. 2009. Retrotransposon expression as a defining event of genome reprograming in fertilized and cloned bovine embryos. Reproduction. 138(2):289–299. doi: 10.1530/REP-09-0042.
- Carmell MA, Girard A, van de Kant HJG, Bourc’his D, Bestor TH, de Rooij DG, Hannon GJ. 2007. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell. 12(4):503–514. doi: 10.1016/j.devcel.2007.03.001.
- Costes V, Chaulot-Talmon A, Sellem E, Perrier J-P, Aubert-Frambourg A, Jouneau L, Pontlevoy C, Hozé C, Fritz S, Boussaha M, et al. 2022. Predicting male fertility from the sperm methylome: application to 120 bulls with hundreds of artificial insemination records. Clin Epigenetics. 14(1):54. doi: 10.1186/s13148-022-01275-x.
- Cui L, Fang L, Shi B, Qiu S, Ye Y. 2018. Spermatozoa expression of piR-31704, piR-39888, and piR-40349 and their correlation to sperm concentration and fertilization rate after ICSI. Reprod Sci. 25(5):733–739. doi: 10.1177/1933719117725822.
- Czech B, Hannon GJ. 2016. One loop to rule them all: the ping-pong cycle and piRNA-guided silencing. Trends Biochem Sci. 41(4):324–337. doi: 10.1016/j.tibs.2015.12.008.
- Donkin I, Barrès R. 2018. Sperm epigenetics and influence of environmental factors. Mol Metab. 14:1–11. doi: 10.1016/j.molmet.2018.02.006.
- Fu Q, Wang PJ. 2014. Mammalian piRNAs: biogenesis, function, and mysteries. Spermatogenesis. 4(1):e27889. doi: 10.4161/spmg.27889.
- Gong J, Wang P, Liu J-C, Li J, Zeng Q-X, Yang C, Li Y, Yu D, Cao D, Duan Y-G. 2022. Integrative analysis of small RNA and mRNA expression profiles identifies signatures associated with chronic epididymitis. Front Immunol. 13:883803. doi: 10.3389/fimmu.2022.883803.
- Gou L-T, Dai P, Yang J-H, Xue Y, Hu Y-P, Zhou Y, Kang J-Y, Wang X, Li H, Hua M-M, et al. 2014. Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell Res. 24(6):680–700. doi: 10.1038/cr.2014.41.
- Han BW, Zamore PD. 2014. piRNAs. Curr Biol. 24(16):R730–R733. doi: 10.1016/j.cub.2014.07.037.
- He L, Wu X, Wu R, Guo P, He W, Sun W, Chen H. 2022. Seminal plasma piRNA array analysis and identification of possible biomarker piRNAs for the diagnosis of asthenozoospermia. Exp Ther Med. 23(5):347. doi: 10.3892/etm.2022.11275.
- Hong Y, Wang C, Fu Z, Liang H, Zhang S, Lu M, Sun W, Ye C, Zhang C-Y, Zen K, et al. 2016. Systematic characterization of seminal plasma piRNAs as molecular biomarkers for male infertility. Sci Rep. 6(1):24229. doi: 10.1038/srep24229.
- Kelleher ES, Barbash DA. 2013. Analysis of piRNA-mediated silencing of active TEs in drosophila melanogaster suggests limits on the evolution of host genome defense. Mol Biol Evol. 30(8):1816–1829. doi: 10.1093/molbev/mst081.
- Khan DR, Fournier É, Dufort I, Richard FJ, Singh J, Sirard M-A. 2016. Meta-analysis of gene expression profiles in granulosa cells during folliculogenesis. Reproduction. 151(6):R103–R110. doi: 10.1530/REP-15-0594.
- Lin H, Spradling AC. 1997. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development. 124(12):2463–2476. doi: 10.1242/dev.124.12.2463.
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song J-J, Hammond SM, Joshua-Tor L, Hannon GJ. 2004. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 305:6.
- Loubalova Z, Fulka H, Horvat F, Pasulka J, Malik R, Hirose M, Ogura A, Svoboda P. 2021. Formation of spermatogonia and fertile oocytes in golden hamsters requires piRNAs. Nat Cell Biol. 23(9):992–1001. doi: 10.1038/s41556-021-00746-2.
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12):550. doi: 10.1186/s13059-014-0550-8.
- Miousse IR, Chalbot M-CG, Lumen A, Ferguson A, Kavouras IG, Koturbash I. 2015. Response of transposable elements to environmental stressors. Mutat Res Rev Mutat Res. 765:19–39. doi: 10.1016/j.mrrev.2015.05.003.
- Morgan HD, Santos F, Green K, Dean W, Reik W. 2005. Epigenetic reprogramming in mammals. Hum Mol Genet. 14 Spec No 1(suppl_1):R47–R58. doi: 10.1093/hmg/ddi114.
- Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD. 2019. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet. 20(2):89–108. doi: 10.1038/s41576-018-0073-3.
- Pantano L, Jodar M, Bak M, Ballescà JL, Tommerup N, Oliva R, Vavouri T. 2015. The small RNA content of human sperm reveals pseudogene-derived piRNAs complementary to protein-coding genes. RNA. 21(6):1085–1095. doi: 10.1261/rna.046482.114.
- Robert C, Nieminen J, Dufort I, Gagné D, Grant JR, Cagnone G, Plourde D, Nivet A-L, Fournier É, Paquet É, et al. 2011. Combining resources to obtain a comprehensive survey of the bovine embryo transcriptome through deep sequencing and microarrays. Mol Reprod Dev. 78(9):651–664. doi: 10.1002/mrd.21364.
- Roovers EF, Rosenkranz D, Mahdipour M, Han C-T, He N, Chuva de Sousa Lopes SM, van der Westerlaken LAJ, Zischler H, Butter F, Roelen BAJ, et al. 2015. Piwi proteins and piRNAs in mammalian oocytes and early embryos. Cell Rep. 10(12):2069–2082. doi: 10.1016/j.celrep.2015.02.062.
- Rosenkranz D, Han C-T, Roovers EF, Zischler H, Ketting RF. 2015. Piwi proteins and piRNAs in mammalian oocytes and early embryos: from sample to sequence. Genom Data. 5:309–313. doi: 10.1016/j.gdata.2015.06.026.
- Rosenkranz D, Zischler H, Gebert D. 2022. piRNAclusterDB 2.0: update and expansion of the piRNA cluster database. Nucleic Acids Res. 50(D1):D259–D264. doi: 10.1093/nar/gkab622.
- Russell SJ, Patel M, Gilchrist G, Stalker L, Gillis D, Rosenkranz D, LaMarre J. 2017. Bovine piRNA-like RNAs are associated with both transposable elements and mRNAs. Reproduction. 153(3):305–318. doi: 10.1530/REP-16-0620.
- Russell SJ, Stalker L, LaMarre J. 2017. PIWIs, piRNAs and retrotransposons: complex battles during reprogramming in gametes and early embryos. Reprod Domest Anim. 52 Suppl 4(S4):28–38. doi: 10.1111/rda.13053.
- Saacke RG. 2008. Sperm morphology: its relevance to compensable and uncompensable traits in semen. Theriogenology. 70(3):473–478. doi: 10.1016/j.theriogenology.2008.04.012.
- Sellem E, Jammes H, Schibler L. 2021. Sperm-borne sncRNAs: potential biomarkers for semen fertility? Reprod Fertil Dev. 34(2):160–173. doi: 10.1071/RD21276.
- Sellem E, Marthey S, Rau A, Jouneau L, Bonnet A, Perrier J-P, Fritz S, Le Danvic C, Boussaha M, Kiefer H, et al. 2020. A comprehensive overview of bull sperm-borne small non-coding RNAs and their diversity across breeds. Epigenetics Chromatin. 13(1):1–28. doi: 10.1186/s13072-020-00340-0.
- Senft AD, Macfarlan TS. 2021. Transposable elements shape the evolution of mammalian development. Nat Rev Genet. 22(11):691–711. doi: 10.1038/s41576-021-00385-1.
- Sharma U. 2019. Paternal contributions to offspring health: role of sperm small RNAs in intergenerational transmission of epigenetic information. Front Cell Dev Biol. 7:215. doi: 10.3389/fcell.2019.00215.
- Utt MD. 2016. Prediction of bull fertility. Anim Reprod Sci. 169:37–44. doi: 10.1016/j.anireprosci.2015.12.011.
- Werry N, Russell SJ, Gillis DJ, Miller S, Hickey K, Larmer S, Lohuis M, Librach C, LaMarre J. 2022. Characteristics of miRNAs present in bovine sperm and associations with differences in fertility. Front Endocrinol. 13:874371. doi: 10.3389/fendo.2022.874371.