
ABSTRACT
Fusion of proteins to the Fc region of IgG is widely used to express cellular receptors and other extracellular proteins, but cleavage of the fusion partner is sometimes required for downstream applications. Immunoglobulin G-degrading enzyme of Streptococcus pyogenes (IdeS) is a protease with exquisite specificity for human IgG, and it can also cleave Fc-fusion proteins at a single site in the N-terminal region of the CH2 domain. However, the site of IdeS cleavage results in the disulfide-linked hinge region partitioning with the released protein, complicating downstream usage of the cleaved product. To tailor the Fc fragment for release of partner proteins by IdeS treatment, we investigated the effect of deleting regions of IgG-derived sequence that are upstream of the cleavage site. Elimination of the IgG-derived hinge sequence along with several residues of the CH2 domain had negligible effects on expression and purity of the fusion protein, while retaining efficient processing by IdeS. An optimal Fc fragment comprising residues 235–447 of the human IgG1 heavy chain sufficed for efficient production of fusion proteins and minimized the amount of residual Ig-derived sequence on the cleavage product following IdeS treatment. Pairing of this truncated Fc fragment with IdeS cleavage enables highly specific cleavage of Fc-fusion proteins, thus eliminating the need to engineer extraneous cleavage sequences. This system should be helpful for producing Fc-fusion proteins requiring downstream cleavage, particularly those that are sensitive to internal miscleavage if treated with alternative proteases.
Abbreviations
| EPOR | = | erythropoietin receptor |
| Fc | = | fragment crystallizable |
| IdeS | = | Immunoglobulin G-degrading enzyme of Streptococcus pyogenes |
| mAb | = | monoclonal antibody |
| PBS | = | phosphate-buffered saline |
| SEC | = | size exclusion chromatography |
| scFv | = | single chain variable fragment |
| TRAILR2 | = | TNF-related apoptosis inducing ligand receptor 2 |
Introduction
Fusion of the extracellular regions of cell surface proteins to the Fc region of IgG is a common method for the recombinant production of these molecules. Such proteins have found widespread use as research and diagnostic reagents.Citation1-3 The Fc region imparts a number of advantages, including enhancement of expression level, simple purification by affinity chromatography, and sensitive detection in immunoassays with Fc-specific reagents. The bivalent nature can also increase the activity of the partner protein if avidity contributes to higher affinity binding. Fusion to Fc is also advantageous for in vivo studies because the Fc region prolongs the serum half-life via interaction with the neonatal Fc receptor, and can elicit effector functions mediated by engagement of other Fc receptors. For these reasons, the application of Fc fusion technology has also been applied to the generation of biotherapeutic drugs, such as etanercept and abatercept.Citation4,5
Although the Fc region confers many useful properties, its presence can complicate the in vitro characterization of a partner protein. For instance, the bivalent nature of Fc-fusion proteins can distort assays that attempt to measure affinity or activity. It can also interfere with structural studies, where the presence of an Fc region is likely to prevent efforts to crystallize a protein of interest. For these reasons, a variety of systems have been described for cleavage of Fc-fusion proteins to release the protein of interest. These have ranged from the use of partial papain proteolysis,Citation6 a protease used to generate Fab fragments from IgG molecules, to the insertion of specific cleavage sites between the partner protein and Fc region that can be recognized by proteases with restricted specificity.Citation7-9 While these approaches are perfectly viable, they usually require insertion of the recognition sequence between the Fc and partner protein, often with additional spacer sequence to enable efficient access of the protease to the cleavage site. Much of this added sequence remains appended to the partner protein after cleavage, and off-target cleavages may sometimes occur if there are internal sequences that resemble the recognition motif.
Immunoglobulin G-degrading enzyme of Streptococcus pyogenes (IdeS) is a highly specific cysteine protease that cleaves human IgG molecules at a single site just below the hinge sequence. IdeS is unusual in its extremely restricted substrate specificity for IgG. No other naturally occurring protein or synthetic peptide substrate has been identified for IdeS.Citation10 All 4 human IgG subtypes can be cleaved (IgG1, IgG2, IgG3, IgG4), but not antibodies of other isotypes. In the case of human IgG1, cleavage occurs between a pair of glycine residues in the N-terminal region of the CH2 domain.Citation11 Peptide fragments encompassing the natural cleavage site in human antibodies are not cleaved by IdeS, but it will digest a papain fragment of human IgG that contains an intact Fc region and 12–15 residues of sequence upstream from the cleavage site.Citation10 These data indicate that a folded Fc region is necessary for substrate recognition by IdeS, but the exact requirements for IgG-derived sequence upstream of the cleavage site remain undefined.
The highly specific cleavage of IgG molecules by IdeS led us to consider whether this protease could be used as a tool to cleave recombinant Fc-fusion proteins. Several examples of Fc-fusion proteins, where the Fc was derived from human IgG1, have indeed been reported to be cleaved by IdeS.Citation12-14 In each of these examples, the Fc partner contained the entire hinge region, as is typical for Fc-fusion proteins. The presence of the IgG hinge region complicates the practical usage of proteins released by IdeS cleavage, as the cleaved product retains this sequence and forms F(ab′)2-like disulfide-linked homodimers. Ideally, if IdeS were to be used as a tool for releasing monomeric proteins of interest from an Fc fusion, the design of the Fc fragment should be optimized in an effort to eliminate unnecessary IgG sequence upstream of the IdeS cleavage site.
Here, we explored the sequence requirements necessary for cleavage of Fc-fusion proteins by IdeS. Elimination of the hinge sequence and the first few residues of the CH2 domain did not affect the production of fusion proteins, or the effectiveness of IdeS cleavage. Based on these results, we designed a truncated Fc fragment that can be used to produce Fc-fusion proteins that are efficiently cleaved by IdeS to release partner proteins as monomeric species with almost no residual fusion sequence at the C-terminus.
Results
Cleavage of Fc-fusion proteins by IdeS
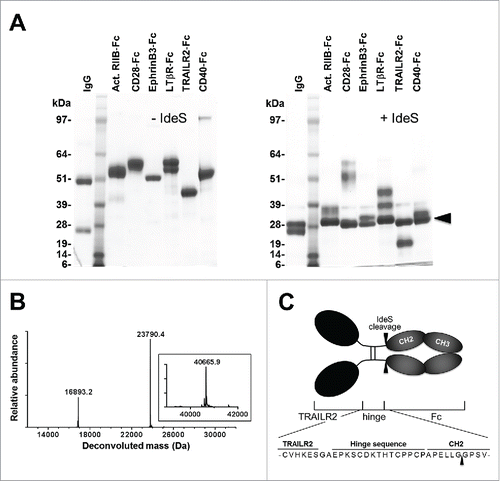
Our initial interest in evaluating IdeS for cleavage of Fc-fusion proteins stemmed from a desire to generate a monomeric fragment of TNF-related apoptosis inducing ligand receptor 2 (TRAILR2) that would be suitable for structural studies. We initially evaluated thrombin for this purpose, by inserting a thrombin recognition site between the TRAILR2 partner protein and Fc region as described by others.Citation8,15 However, in addition to cleavage at this designed site, we unexpectedly found that thrombin also cleaved TRAILR2 at an internal arginine residue within the motif -PQQR^AA- (data not shown). IdeS has been shown to cleave some Fc-fusion proteins,Citation12-14 so we decided to investigate this in the context of our protein of interest. To confirm the general ability of IdeS to specifically cleave proteins fused to human IgG1-Fc, we treated TRAILR2-Fc and a broader panel of Fc-fusion proteins with IdeS. In all cases, IdeS treatment resulted in efficient cleavage of the fusion construct and release of the partner protein (). Mass spectrometry was used to determine the cleavage site within the TRAILR2-Fc-fusion protein, and this analysis confirmed that cleavage of this fusion protein occurred at the anticipated (-Gly^Gly-) site within the Fc region ().
Figure 1. Cleavage of Fc-fusion proteins by IdeS. (A) Reducing SDS-PAGE analysis of uncleaved Fc-fusion proteins (left panel) and following treatment with IdeS (right panel). Lanes were loaded with the following samples: 1) total human IgG; 2) molecular weight standards; 3) Activin RIIB-Fc; 4) CD28-Fc; 5) EphrinB3-Fc; 6) Lymphotoxin βR-Fc; 7) TRAILR2-Fc ; 8) CD40-Fc. All samples were commercially obtained except for TRAILR2-Fc and CD40-Fc which were in-house reagents. The arrowhead in the right panel marks migration of the cleaved Fc fragment. (B) Intact mass spectra of IdeS-treated TRAILR2-Fc after deglycosylation and reduction. The experimentally observed masses were in good agreement with the calculated masses of fragments based on cleavage between Gly236-Gly237 in the Fc region (calculated TRAILR2-containing fragment mass: 16,892.9 Da; calculated Fc fragment mass: 23,791.9 Da). The inset shows the intact mass of untreated TRAILR2-Fc following deglycosylation and reduction (calculated mass: 40,666.8 Da). (C) Schematic representation of the TRAILR2-Fc-fusion protein and location of the IdeS cleavage site.
Design and cleavage of fusion proteins containing truncated Fc fragments
As shown in , cleavage of TRAILR2-Fc by IdeS resulted in release of the TRAILR2 partner protein, but the molecule retained a 23-residue fragment at the C-terminus derived from the hinge and CH2 regions of human IgG1 heavy chain. In addition to being a long C-terminal appendage, this segment also contained cysteine residues that are expected to form interchain disulfide bonds. To determine if this part of the IgG1-derived sequence could be minimized without effecting expression of the fusion protein or its cleavage by IdeS, a series of modified TRAILR2-Fc fusion constructs were designed that lacked the hinge sequence and N-terminal residues of the CH2 domain. One construct, TRAILR2-Fc1, lacked the genetically-encoded hinge region of IgG1, while additional constructs TRAILR2-Fc2, -Fc3 and Fc4 lacked the hinge region and portions of the CH2 domain that reside upstream of the IdeS cleavage site ().
Figure 2. Expression, purification and characterization of TRAILR2-Fc variants. (A) Design of fusion protein variants. Gray sequence represents the boundaries of TRAILR2 and IgG1 sequence that were common to all constructs. The region of sequence that varied between constructs is shown in black and was derived from the hinge and N-terminal region of the CH2 domain. Lower case sequences were additional residues in TRAILR2-Fc and TRAILR2-Fc1 that resulted from the cloning procedure. An asterisk marks the anticipated IdeS cleavage site. (B) Non-reducing SDS-PAGE analysis of purified TRAILR2-Fc (TR2-Fc) variants; lanes 1–4 = TRAILR2-Fc1-Fc4 respectively and lane 5 = TRAILR2-Fc. (C) SEC analysis of purified TRAILR2-Fc and TRAILR2-Fc3 fusion proteins showing near identical retention times. Remaining constructs had similar chromatographic profiles and retention times.
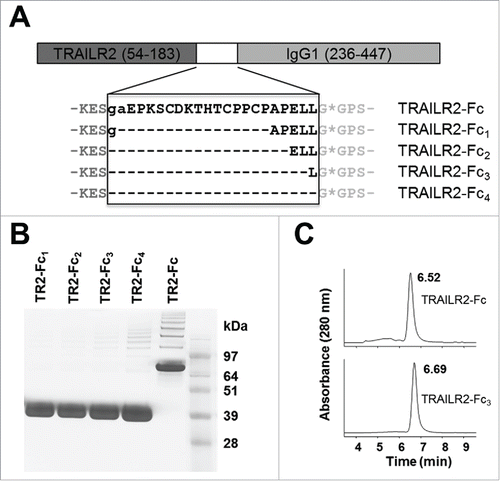
The parental and modified TRAILR2-Fc-fusion proteins were expressed in a 293F transient expression system and purified from culture media by protein A affinity chromatography. In this transient system, the expression level was similar for all constructs, between 100–150 mg/L based on the yield of purified material. Samples were assessed by non-reducing SDS-PAGE and size-exclusion chromatography (SEC) to determine if each was a covalent or noncovalent dimer. Non-reducing SDS-PAGE analysis () showed that the parental fusion construct ran as a disulfide-linked dimer, as expected for this protein given the presence of interchain disulfide bonds within the hinge region. By contrast, the modified fusions ran as monomers on non-reducing SDS-PAGE, consistent with the absence of the IgG1 hinge region in these constructs. Analytical SEC was used to assess the oligomerization status under native conditions. Each of the modified fusion constructs exhibited similar retention times as parental TRAILR2-Fc when analyzed by SEC, suggesting they all formed Fc-mediated homodimers (). This showed that noncovalent forces were sufficient to maintain homodimerization of the Fc region in the absence of interchain disulfide bonds. Interestingly, constructs that lacked the hinge region were also of higher purity in that they contained lower levels of higher order aggregated material, as observed by SEC.
IdeS specificity is largely independent of upstream IgG sequence
Each of the purified fusion proteins was treated with IdeS to determine their susceptibility to specific cleavage. Samples of each construct at a concentration of 2 mg/mL were treated with 1U IdeS/μg of substrate (2U/μL) for 1 hr at 37°C, prior to quenching the reaction and analyzing by SDS-PAGE (). With the exception of construct TRAILR2-Fc4, each of the fusion protein samples was >90% cleaved by IdeS under these conditions. Elimination of upstream IgG1-derived sequence did not alter the site of IdeS cleavage between Gly236-Gly237, as shown by mass spectrometry for cleavage of TRAILR2-Fc3 (). Partial cleavage of TRAILR2-Fc4 suggested that a leucine in the P2 positionCitation16 of an Fc fusion substrate may be an important determinant of IdeS specificity. Thus, the upstream specificity requirements for processing by IdeS were largely restricted to retention of Leu235 in the P2 position of substrate, and possibly Gly236 in P1. This is consistent with structural modeling of substrate binding to IdeSCitation17 and less efficient digestion of human IgG2 antibodies that contain a Val-Ala in the equivalent positions.Citation11
Figure 3. IdeS cleavage of TRAILR2-Fc variants and limited proteolysis of TRAILR2-Fc3. (A) Reducing SDS-PAGE of IdeS-treated TRAILR2-Fc (TR2-Fc) and variant constructs. Lanes 2 – 5 correspond to TRAILR2-Fc1 - Fc4 samples after treatment with IdeS for 1 h. Lane 6 is the parental TRAILR2-Fc construct treated in the same way. Sample loadings were 10 μg of protein per lane. (B) Intact mass spectra of IdeS-treated TRAILR2-Fc3 after deglycosylation and reduction. The experimentally observed masses were in good agreement with the calculated masses of fragments based on cleavage between Gly236-Gly237 in the Fc region (calculated TRAILR2 fragment mass: 14,729.5 Da; calculated Fc fragment mass: 23,791.9 Da). The inset shows the intact mass of untreated TRAILR2-Fc following deglycosylation and reduction (calculated mass: 38503.4 Da). (C) Reducing SDS-PAGE (10% Bis-Tris gel) analysis of TRAILR2-Fc3 digestion as a function of time and IdeS concentration. Concentration of TRAILR2-Fc3 was 2 mg/mL (52 uM) in all reactions, and sample loadings were 5 μg of protein per lane.
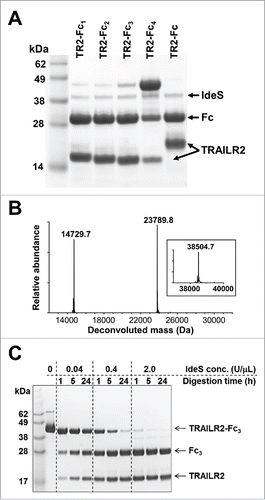
Our digestion conditions had followed the manufacturer's recommendation of using 1 U of IdeS per microgram of substrate, which we have estimated to represent a protease:substrate ratio of 1:100 w/w (see Materials and Methods). While this is comparable to the ratios used with other site-specific proteases, an ability to reduce the amount of IdeS needed would help to make use of this reagent more affordable, particularly if a large amount of fusion protein is to be cleaved. Accordingly, for a fixed substrate concentration of 2 mg/mL TRAILR2-Fc3 (52 μM), we evaluated cleavage as a function of IdeS concentration and incubation time at 37°C. As shown in , cleavage of TRAILR2-Fc was essentially complete within 1 h when treated with 2U/μL (i.e., 1U/μg) of IdeS, but incomplete at concentrations 5 or 50-fold below this. However, longer incubation times could partially compensate for lower IdeS concentrations, and near complete cleavage of TRAILR2-Fc3 was achieved by incubation with 0.4 U/μL IdeS for 24 h at 37°C. The TRAILR2 protein released by cleavage of TRAILR2-Fc3 under these conditions was readily purified using a mixture of protein A and Ni-affinity resins to remove the Fc fragment, residual uncleaved fusion protein and the His-tagged IdeS.
General application to production and cleavage of Fc-fusion proteins
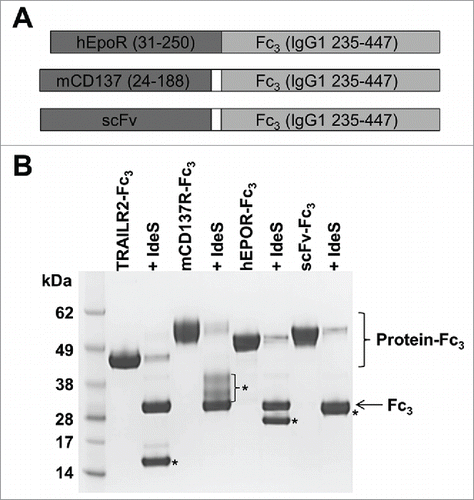
The design of the TRAILR2-Fc3 construct combined rapid cleavage by IdeS with a minimal level of residual IgG1 sequence on the released TRAILR2 protein. As such, IgG1 235–447 appeared to be an optimal fragment for use in fusions proteins that are cleaved with IdeS. To validate that this fragment would be generally useful in producing and cleaving different Fc-fusion proteins, we expressed 3 additional proteins using this system. Constructs encoding the extracellular regions of human erythropoietin receptor (EPOR), mouse CD137, and a single-chain variable fragment (scFv) fused to IgG1 235–447 were generated () and used to transiently transfect HEK293F cells. Each of these fusion proteins expressed in reasonable yields and were readily purified by protein A affinity chromatography. Together with TRAILR2-Fc3 as a positive control, 52 μM samples of each protein were incubated with 2 U/μL IdeS for 1 h prior to quenching the reaction and analyzing by SDS-PAGE. Each of these proteins was >90% cleaved under these standard reaction conditions (). Digestion at a single site between residues Gly236 and Gly237 (IgG1 numbering) was confirmed for each of these fusion proteins by mass spectrometry (data not shown), thereby demonstrating the general applicability of this system for producing and cleaving fusions to different proteins.
Figure 4. Design and IdeS cleavage of proteins fused to IgG1 235–447. (A) Design of fusion proteins. Extracellular regions of human EPOR and mouse CD137, or an scFv protein were fused to an IgG1 235–447 Fc fragment. The EPOR fragment was a direct fusion to IgG1 235–447, while CD137 and scFv fusions contained a 2 residue Gly-Ser or Gly-Gly linker between Fc and the upstream protein. (B) Reducing SDS-PAGE (10% Bis-Tris gel) of fusion proteins before and after treatment with 2 U/μL IdeS for 1 h at 37°C. IdeS-treated samples appear in the lane immediately to the right of the untreated sample. Migration of the cleaved Fc3 fragment is shown by an arrow, while the various partner protein fragments are marked by asterisks. The CD137 fragment contains up to 2 N-linked glycans and runs as a diffuse band, while the scFv band is partially overlapping with the Fc3 fragment.
IdeS can also cleave fusions to an isolated CH2 domain
Production of Fc-fusion proteins is generally restricted to eukaryotic expression hosts, limiting the potential use of bacterial expression systems to produce fusions that can be cleaved by IdeS. While it may not offer equivalent advantages of the full Fc as a fusion partner, the CH2 domain subfragment of Fc can be produced in E. coli at high levels,Citation18 and could be used as a fusion partner for bacterially expressed proteins. However, the ability of IdeS to cleave fusions to this single domain has not been previously explored. To evaluate this, we expressed the CH2 module of human IgG1 in E. coli, with an N-terminal tag-containing peptide extension as a surrogate for a potential partner protein (). This module was expressed in soluble form intracellularly in E. coli, and purified via a His-tag present in the N-terminal extension (). Material obtained in this manner was mostly in a reduced state as assessed by mass spectrometry and reverse phase HPLC, but spontaneous formation of the intramolecular disulfide bond occurred after room temperature storage for several days at pH 8.3.
Figure 5. IdeS cleavage of a CH2-peptide fusion protein. (A) Amino acid sequence of the CH2-peptide fusion protein. The CH2 domain sequence contained an N-terminal peptide extension (underlined) with a 6x histidine tag and additional vector derived sequence. The terminal methionine residue is shown in gray to reflect processing of this residue that occurs during bacterial expression. The predicted IdeS cleavage site is denoted by an asterisk. (B) Reducing SDS-PAGE (4–12% Bis-Tris gel) of purified TRAILR2-Fc3 and CH2-peptide fusion proteins (lanes 2 and 4 respectively), and after treatment of 52 μM samples with 2 U/μL IdeS at 37°C (lanes 3, 5 and 6). IdeS-treated samples appear in lanes to the right of the untreated sample, and the IdeS incubation times are noted in parentheses. In contrast to TRAILR2-Fc3 cleavage (lane 3), the CH2-peptide fusion was only partially cleaved after a 1 h incubation with IdeS (lane 5). However, extending the incubation time to 16 h under otherwise identical cleavage conditions led to near complete cleavage of the CH2-peptide fusion (lane 6).
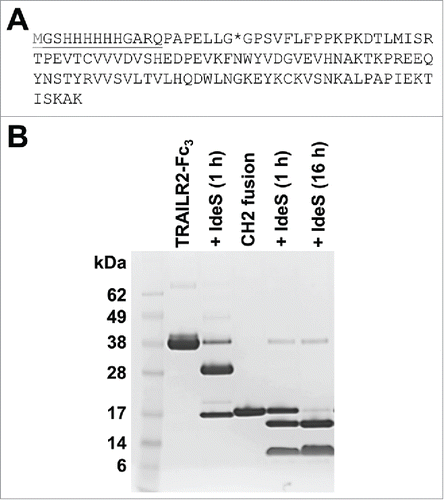
Incubation of the recombinant CH2 with IdeS, using equivalent molar concentrations of substrate and enzyme as previously used for TRAILR2-Fc3, resulted in specific cleavage of the CH2 domain fusion, albeit at a slower rate than for cleavage of Fc-fusion proteins (). However, extending the IdeS incubation time from 1 h to 16 h resulted in complete cleavage of the CH2 fusion protein. Liquid chromatography/mass spectrometry analysis of the non-reduced digestion mixture identified 2 products of molecular weight 11,826 Da and 2,075 Da derived from the 13,883 Da starting material. These product masses corresponded to specific cleavage at the expected Gly-Gly peptide bond near the N-terminus of the CH2 domain. Although cleavage was less efficient than for Fc-fusion proteins, this shows that site-specific cleavage of a bacterially-expressed CH2-fusion protein by IdeS could be feasible if there was a desire to use this protease in conjunction with a bacterial expression system.
Discussion
The highly restricted specificity of IdeS makes it the ultimate site-specific protease for cleavage of human IgGs, as well as Fc-fusion proteins. This has led to its widespread use for analytical characterization of these molecules,Citation12,14,19 but its utility in providing monomeric products from digested Fc-fusion proteins is handicapped by the cleavage below the disulfide-containing hinge region. In an effort to tailor the Fc fragment for optimal pairing with IdeS, we found that a truncated fragment lacking the hinge sequence and proximal CH2 residues is a perfectly adequate fusion partner for expression of extracellular proteins. Use of the truncated fragment did not compromise the yield or quality of fusion products, and cleavage by IdeS was highly specific and afforded partner proteins that were free of all but 2 residues of Ig-derived sequence. Fusions containing this truncated Fc region are unlikely to retain effector functions because Fcγ receptor binding is known to involve the N-terminal region of the CH2 domain.Citation20 This is generally unimportant for in vitro applications; however, if retention of Fcγ receptor binding were required, it may be possible to restore this function by adding back some or all of the N-terminal CH2 residues.
The current practice of using Fc fragments that incorporate the entire hinge sequence has a historical basis, but not an experimental one where this was deemed most effective. The earliest examples of using the Fc fragment as a fusion partner were in expression of the ligand-binding domains of CD4.Citation21,22 Since then, most if not all Fc-fusion proteins have utilized an Fc fragment with hinge-CH2-CH3 configuration, most typically derived from human IgG1. This design is convenient in maintaining the hinge as a natural spacer, though as we show here, this is not a requirement for effective expression of Fc-fusion proteins. In addition to eliminating the hinge, we also showed that the first several residues of the CH2 domain can be deleted. It's important to note that the sequence boundaries of the CH2 domain are defined on the basis of an exon-encoded segment in the IgHG gene, and not on structural considerations. The first 6–8 residues of the CH2 domain in human IgG1 are not part of the structural immunoglobulin domain,Citation23,24 and indeed this terminal region of CH2 is frequently referred to as the “lower hinge”.Citation25,26 In the context of an isolated CH2 domain from human IgG1, deletion of the 7 N-terminal residues (Ala231 – Gly237) actually improved the structural stability of this domain. Taken together, it is therefore unsurprising that the sequence upstream of Gly237 can be dispensed for production of Fc-fusion proteins, though its inclusion can be useful as a native sequence spacer between partner protein and Fc.
Most proteases can digest synthetic peptides that encompass a cleavage site within a protein substrate, but IdeS is unusual in showing no detectable digestion of linear peptides that span the natural cleavage sites in human IgG.Citation10 This indicates that IdeS will only digest substrates containing a folded Fc-region, and the current work shows that cleavage is largely independent of the sequence or structure of the substrate upstream of the cleavage site. This unique specificity has been attributed to an exosite in IdeS that is involved in recognition of the Fc region.Citation10,17 Our observation that a tagged CH2 domain protein can also be processed suggests that this is the minimal folded unit required for IdeS recognition, but additional features of the full Fc may enhance productive binding to IdeS given the slower rate of cleavage for CH2 versus Fc fusion molecules.
As shown by others, several of the commonly used site-specific proteases have been used to cleave partner proteins from an Fc fusion by insertion of the relevant recognition sequence between these 2 regions. However, IdeS recognizes the native Fc sequence, thereby obviating the need for a foreign cleavage motif. The system described here may have additional advantages over alternative proteases depending on the properties or downstream requirements for the cleaved product. For instance, IdeS is extraordinarily specific, so for partner proteins that are susceptible to internal cleavages with an alternative protease, as was the case for miscleavage of TRAILR2 by thrombin, use of IdeS would be preferred. Another benefit, relative to other cysteine proteases, such as papain and TEV protease, is that IdeS cleavage buffers do not require reducing agents that can otherwise disrupt labile disulfide bonds. Finally, most site-specific proteases cleave at or near the C-terminus of their recognition motifs, meaning that all or most of that motif remains appended after cleavage of a C-terminal fusion partner (e.g., 4 residues in the case of thrombin, 6 residues for TEV protease). We demonstrated rapid cleavage of Fc-fusion proteins to generate cleavage products with only 2 residues of residual fusion partner sequence, and it could be possible to reduce this further as demonstrated by cleavage of the TRAILR2-Fc4 construct, albeit at the expense of cleavage efficiency. While this may seem like a modest advantage, unstructured regions can inhibit protein crystallization or the quality of diffraction data, so any reduction in the amount of residual fusion partner sequence could be of benefit for crystallographic applications.
In conclusion, we characterized the sequence requirements for effective cleavage of Fc-fusion proteins by IdeS, and developed a method for using this protease to release monomeric cleavage products, in a near traceless manner, by truncating the Fc fragment used as a fusion partner. We think that use of this IdeS-Fc fusion system may be preferable to other cleavable Fc-fusion proteins in certain circumstances, and will further enhance the utility of this class of molecule for scientific applications.
Materials and methods
Reagents
Recombinant Fc-fusion proteins were either produced in house (human TRAILR2, CD40, EPOR, murine CD137, anti-TRAILR2 scFv-Fc) or were purchased from R&D Systems (human Activin RIIB, CD28, Ephrin-B3 and lymphotoxin βR). In all cases, the Fc regions were derived from human IgG1. IdeS protease (tradename “FabRICATOR”) was purchased from Genovis. All other biochemicals were from Sigma-Aldrich unless otherwise noted.
Cleavage conditions and methods of analysis
Unless otherwise indicated, 2 mg/mL samples of fusion proteins in 50 mM sodium phosphate buffer, 150 mM sodium chloride, pH 6.6 were treated at 37°C with 1 U IdeS/μg substrate (i.e. 2 U IdeS/μL). To quench cleavage reactions at predefined time points, samples were acidified by addition of 2 μL of 3 M hydrochloric acid per 15 μL of reaction aliquot. After 5 min incubation at room temperature (to irreversibly denature IdeS), samples were neutralized by addition of 1 μL 5 M sodium hydroxide and 3 μL of 1 M sodium phosphate pH 7.0, then frozen at −80°C prior to SDS-PAGE analysis. While the manufacturer does not define a mass equivalent for 1 U of IdeS, we could typically detect a faint IdeS band on stained gels, near the limit of detection, after loading the equivalent of 5 U of enzyme. From this, we estimate 5 U of IdeS contains ∼50 ng of protein (i.e., 1 U IdeS is ∼10 ng). Accordingly, we estimate the IdeS:substrate ratio was ∼1:100 w/w under these standard cleavage conditions.
Unless stated otherwise, SDS-PAGE analysis was conducted with precast gels (Invitrogen) using the manufacturers sample and running buffers. For reduction of samples prior to gel analysis, dithiothreitol was added to a final concentration of 50 mM prior to loading of samples onto the gel. For analysis of cleavage reactions by mass spectrometry, samples were deglycosylated and reduced prior to liquid chromatography-mass spectrometry. Typically, reaction samples containing 30 μg of protein were incubated with 1 μL Rapid™ PNGase F (New England Biolabs) for 10 minutes according to the manufacturer's protocol, then reduced by addition of dithiothreitol to a final concentration of 50 mM. Liquid chromatography-mass spectrometry analysis was performed with an Agilent 6520B Q-TOF mass spectrometer connected in-line to a 1200 series HPLC equipped with a reverse phase column (Agilent Poroshell 300SB-C3; 5 um, 2.1 mm × 75 mm). Samples were injected onto the column using a mobile phase of 10% v/v aqueous acetonitrile containing 0.1% v/v formic acid. After a brief isocratic hold to allow for sample desalting, proteins were eluted by a change in mobile phase to 60% v/v aqueous acetonitrile, 0.1% v/v formic acid. Mass data were collected for 100–3000 m/z, in positive polarity mode using a gas temperature of 350°C, nebulizer pressure of 48 psi and capillary voltage of 5000 V. Data were analyzed using the vendor-supplied software (Agilent MassHunter Qualitative Analysis v.B.04.00).
Expression and purification of Fc-fusion proteins
Synthetic gene sequences and oligonucleotides were ordered from GeneArt (Thermo Fisher Scientific), or Integrated DNA Technologies. A synthetic gene encoding amino acids 54–183 of human TRAILR2 was cloned into a mammalian expression construct containing an upstream signal sequence, cytomegalovirus promoter element, and downstream fragment encoding the hinge-CH2-CH3 region of human IgG1 heavy chain, corresponding to residues 216–447 of IgG1 (residue numbering for IgG1 throughout this work is according to the Eu systemCitation27). This TRAILR2-Fc construct encoded a Gly-Ala dipeptide insertion between the TRAILR2 and Fc fragments resulting from use of a KasI cloning site for introduction of the TRAILR2 sequence. Modified forms of this fusion construct lacking the hinge region (TRAILR2-Fc1) or hinge and N-terminal portions of the CH2 domain (TRAILR2-Fc2, -Fc3 and -Fc4) were generated by using short synthetic duplex DNA cassettes to replace an NcoI – ApaI fragment in the TRAILR2-Fc construct, that fragment encoding the region from Pro172 of TRAILR2 to Pro238 of IgG1. For preparation of other fusion proteins described in this work, synthetic genes encoding the extracellular regions of human EPOR, murine CD137 and an anti-TRAILR2 scFv fused to residues 235–447 of human IgG1 were generated by GeneArt and used to replace the TRAILR2-Fc fragment in the parental construct.
Recombinant expression of Fc-fusion proteins was performed by transient transfection of HEK293F cells with plasmids using 293fectin™ in serum-free Freestyle™ medium according to the supplier's recommended procedures (Invitrogen). Cell culture supernatants were harvested 6 d after transfection, filtered through a 0.22 μm sterile filter, and the fusion proteins were purified using HiTrap MabSelect columns (GE Healthcare) according to the manufacturers' protocol. Purified proteins were characterized by SDS-PAGE and SEC using a Bio-SEC-5 300Å 7.8 mm X 150 mm column (Agilent Technologies) at 1.25 mL/min in PBS running buffer. Analysis of purified fusion proteins by mass spectrometry was performed as described for analysis of IdeS cleavage reactions. Small scale purification of the TRAILR2 fragment released by cleavage of TRAILR2-Fc3 with IdeS was performed by incubating 250 μL of the reaction mixture with 25 μL of MabSelect resin for 1 h at room temperature and removing the resin by centrifugation through an empty micro bio-spin column (Bio-Rad). This process was repeated with 25 μL of Superflow Ni-NTA resin (Qiagen) to remove the His-tagged IdeS protein.
Expression and purification of CH2 domain protein
Residues 230–447 of human IgG1 fused to an N-terminal peptide containing a 6x histidine tag sequence was expressed from a modified pET-15b plasmid in BL21 (DE3) E. coli. Cultures were grown to an OD 600 of 1.0 in MagicMedia™ (Invitrogen) and subsequently chilled on ice prior to incubation at 18°C for 20 h in a shaking incubator. Cells were harvested and lysed using BugBuster® (Novagen) and lysozyme at a concentration of 0.2 mg/mL. Magnesium chloride and DNAse I were added to degrade genomic DNA, and following centrifugation, the clarified lysate was loaded onto a 1 mL HisTrap™ column (GE Healthcare). After washing the column according to the manufacturer protocol, the CH2 protein was eluted using an imidazole gradient in 25 mM Tris-HCl, 300 mM sodium chloride pH 8.3. After several days at room temperature, the eluate was dialyzed against 25 mM sodium phosphate, 25 mM sodium chloride pH 6.6 and purified on a 0.8 mL POROS S column (Applied Biosystems) using a 20 column volume gradient to 600 mM sodium chloride.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Chamow SM, Ashkenazi A. Immunoadhesins: Principles and applications. Trends Biotechnol 1996; 14:52-60; PMID:8746117; http://dx.doi.org/10.1016/0167-7799(96)80921-8
- Brown SJ, Becherer KA, Blumeyer K, Kautzer C, Axelrod F, Le H, McConnell SJ, Whalley A, Spinella DG. Expression and ligand binding assays of soluble cytokine receptor-immunoglobulin fusion proteins. Protein Expr Purif 1998; 14:120-4; PMID:9758759; http://dx.doi.org/10.1006/prep.1998.0940
- Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: New developments and future perspectives. EMBO Mol Med 2012; 4:1015-28; PMID:22837174; http://dx.doi.org/10.1002/emmm.201201379
- Huang C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODY technology. Curr Opin Biotechnol 2009; 20:692-9; PMID:19889530; http://dx.doi.org/10.1016/j.copbio.2009.10.010
- Beck A, Reichert JM. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. MAbs 2011; 3:415-6; PMID:21785279; http://dx.doi.org/10.4161/mabs.3.5.17334
- Chamow SM, Peers DH, Byrn RA, Mulkerrin MG, Harris RJ, Wang WC, Bjorkman PJ, Capon DJ, Ashkenazi A. Enzymatic cleavage of a CD4 immunoadhesin generates crystallizable, biologically active Fd-like fragments. Biochemistry 1990; 29:9885-91; PMID:2125484; http://dx.doi.org/10.1021/bi00494a019
- Beck JT, Marsters SA, Harris RJ, Carter P, Ashkenazi A, Chamow SM. Generation of soluble interleukin-1 receptor from an immunoadhesin by specific cleavage. Mol Immunol 1994; 31:1335-44; PMID:7997245; http://dx.doi.org/10.1016/0161-5890(94)90052-3
- Culouscou JM, Carlton GW, Aruffo A. HER4 receptor activation and phosphorylation of SHC proteins by recombinant heregulin-Fc fusion proteins. J Biol Chem 1995; 270:12857-63; PMID:7759543; http://dx.doi.org/10.1074/jbc.270.21.12857
- Haspel J, Blanco C, Jacob J, Grumet M. System for cleavable Fc fusion proteins using tobacco etch virus (TEV) protease. BioTechniques 2001; 30:60-6; PMID:11196321
- Vincents B, von Pawel-Rammingen U, Bjorck L, Abrahamson M. Enzymatic characterization of the streptococcal endopeptidase, IdeS, reveals that it is a cysteine protease with strict specificity for IgG cleavage due to exosite binding. Biochemistry 2004; 43:15540-9; PMID:15581366; http://dx.doi.org/10.1021/bi048284d
- von Pawel-Rammingen U, Johansson BP, Bjorck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J 2002; 21:1607-15; PMID:11927545; http://dx.doi.org/10.1093/emboj/21.7.1607
- Lynaugh H, Li H, Gong B. Rapid Fc glycosylation analysis of Fc fusions with IdeS and liquid chromatography mass spectrometry. MAbs 2013; 5:641-5; PMID:23839239; http://dx.doi.org/10.4161/mabs.25302
- Strand J, Huang CT, Xu J. Characterization of Fc-fusion protein aggregates derived from extracellular domain disulfide bond rearrangements. J Pharm Sci 2013; 102:441-53; PMID:23242781; http://dx.doi.org/10.1002/jps.23421
- An Y, Zhang Y, Mueller HM, Shameem M, Chen X. A new tool for monoclonal antibody analysis: Application of IdeS proteolysis in IgG domain-specific characterization. MAbs 2014; 6:879-93; PMID:24927271; http://dx.doi.org/10.4161/mabs.28762
- Mine S, Koshiba T, Honjo E, Okamoto T, Tamada T, Maeda Y, Matsukura Y, Horie A, Ishibashi M, Sato M, et al. Thermodynamic analysis of the activation mechanism of the GCSF receptor induced by ligand binding. Biochemistry 2004; 43:2458-64; PMID:14992583; http://dx.doi.org/10.1021/bi0356855
- Schechter I, Berger A. On the size of the active site in proteases. I. papain. Biochem Biophys Res Commun 1967; 27:157-62; PMID:6035483; http://dx.doi.org/10.1016/S0006-291X(67)80055-X
- Wenig K, Chatwell L, von Pawel-Rammingen U, Bjorck L, Huber R, Sondermann P. Structure of the streptococcal endopeptidase IdeS, a cysteine proteinase with strict specificity for IgG. Proc Natl Acad Sci U S A 2004; 101:17371-6; PMID:15574492; http://dx.doi.org/10.1073/pnas.0407965101
- Gong R, Vu BK, Feng Y, Prieto DA, Dyba MA, Walsh JD, Prabakaran P, Veenstra TD, Tarasov SG, Ishima R, et al. Engineered human antibody constant domains with increased stability. J Biol Chem 2009; 284:14203-10; PMID:19307178; http://dx.doi.org/10.1074/jbc.M900769200
- Chevreux G, Tilly N, Bihoreau N. Fast analysis of recombinant monoclonal antibodies using IdeS proteolytic digestion and electrospray mass spectrometry. Anal Biochem 2011; 415:212-4; PMID:21596014; http://dx.doi.org/10.1016/j.ab.2011.04.030
- Caaveiro JM, Kiyoshi M, Tsumoto K. Structural analysis of Fc/FcgammaR complexes: A blueprint for antibody design. Immunol Rev 2015; 268:201-21; PMID:26497522; http://dx.doi.org/10.1111/imr.12365
- Traunecker A, Schneider J, Kiefer H, Karjalainen K. Highly efficient neutralization of HIV with recombinant CD4-immunoglobulin molecules. Nature 1989; 339:68-70; PMID:2541344; http://dx.doi.org/10.1038/339068a0
- Byrn RA, Mordenti J, Lucas C, Smith D, Marsters SA, Johnson JS, Cossum P, Chamow SM, Wurm FM, Gregory T, et al. Biological properties of a CD4 immunoadhesin. Nature 1990; 344:667-70; PMID:1970124; http://dx.doi.org/10.1038/344667a0
- Huber R, Deisenhofer J, Colman PM, Matsushima M, Palm W. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature 1976; 264:415-20; PMID:1004567; http://dx.doi.org/10.1038/264415a0
- Prabakaran P, Vu BK, Gan J, Feng Y, Dimitrov DS, Ji X. Structure of an isolated unglycosylated antibody CH2 domain. Acta Crystallogr D Biol Crystallogr 2008; 64:1062-7; PMID:18931413; http://dx.doi.org/10.1107/S0907444908025274
- Burton DR. Immunoglobulin G: Functional sites. Mol Immunol 1985; 22:161-206; PMID:3889592; http://dx.doi.org/10.1016/0161-5890(85)90151-8
- Feinstein A, Richardson N, Taussig MI. Immunoglobulin flexibility in complement activation. Immunol Today 1986; 7:169-74; PMID:25290202; http://dx.doi.org/10.1016/0167-5699(86)90168-4
- Edelman GM, Cunningham BA, Gall WE, Gottlieb PD, Rutishauser U, Waxdal MJ. The covalent structure of an entire gammaG immunoglobulin molecule. Proc Natl Acad Sci U S A 1969; 63:78-85; PMID:5257969; http://dx.doi.org/10.1073/pnas.63.1.78