
ABSTRACT
Cell surface antigen-specific antibodies are of substantial diagnostic and therapeutic importance. The binding properties of such antibodies are usually evaluated by cell-free assays, in particular surface plasmon resonance (SPR) analysis, or flow cytometry. SPR analyses allow the detailed quantitative and dynamic evaluation of the binding properties of antibodies, but need purified, typically recombinantly produced antigens. It can, however, be difficult to produce the required antigen. Furthermore, cellular factors influencing the antigen-antibody interaction are not considered by this method. Flow cytometry-based analyses do not have these limitations, but require elaborated calibration controls for absolute quantification of bound molecules. To overcome the limitations of SRP and flow cytometry in the characterization of cell surface antigen-specific antibodies, we developed Fn14-specific antibody 18D1 as an example of an antibody fusion protein format that includes the luciferase of Gaussia princeps (GpL), which enables very simple and highly sensitive cellular binding studies. We found that GpL-tagging of the C-terminus of the antibody light chain does not affect the interaction of 18D1-IgG1 with its antigen and Fc-gamma receptors (FcγRs). In accordance with this, the GpL(LC-CT)-18D1-IgG1 antibody fusion protein showed basically the same FcγR-dependent agonistic properties as the parental 18D1 antibody. Similar results were obtained with isotype switch variants of 18D1 and antibodies specific for CD95, LTβR and CD40. In sum, we demonstrate that antibody GpL fusion proteins are easily manageable and versatile tools for the characterization of cell surface antigen-antibody interactions that have the potential to considerably extend the instrumentarium for the evaluation of antibodies.
Abbreviations
| GpL | = | Gaussia princeps luciferase |
| FcγR | = | Fc-gamma receptor |
| Fn14 | = | fibroblast growth factor-inducible 14 |
| TWEAK | = | tumor necrosis factor (TNF)-related weak inducer of apoptosis |
Introduction
Antibodies, particularly those directed against cell surface antigens, are indispensable in diagnosis and therapy of many diseases. Early development and characterization of antigen binding of cell surface-antigen directed antibodies are typically based on the use of cell-free methods, particularly surface plasmon resonance (SPR) analysis and isothermal titration calorimetry.Citation1-2 However, these cell-free methods have 2 severe limitations. First, not only the antibody but also the antigen must be available for use as a soluble analyte or for immobilization on the sensor chip. Therefore, there is typically a need to produce the antigen in a recombinant form. Especially in the case of cell surface antigens, this can be challenging because an appropriate protein domain must be chosen for expression in sufficient quantities, and the functional integrity of the protein domain has to be controlled. In the case of antibodies recognizing a structural epitope composed of 2 different proteins, two independent soluble proteins must be produced, and their correct assembly in solution must be ensured. Second, cellular factors, for example interactions of the cell surface antigen with the cytoskeleton or plasma membrane components or unknown binding partners of the antigen, may modulate the efficacy/dynamics of antibody binding. Both limitations can be straightforwardly overcome by cellular binding studies in which antibody binding to cell expressed cell surface antigens is directly measured. Cellular binding studies, however, are crucially dependent on antibody variants that allow highly sensitive and accurate quantification of the cell-bound antibody molecules. Although there are many examples of biochemical labeling of antibodies with iodine, biotin or fluorogenic compounds, it can be very challenging to find reaction conditions that ensure antibody labeling with a useful efficiency in a reproducible manner and without disturbance of the antigen- or Fcγ receptor (FcγR)-binding properties of the antibody. Moreover, biochemical methods of antibody labeling typically result in mixtures of antibody species that are labeled to a varying degree and on different positions. This heterogeneity can be especially problematic in quantitative functional studies because the antibodies with the highest label load will definitely dominate the calculation of the specific label activity per antibody, but these molecule species also have the highest risk of being affected in their antigen- or FcγR-binding properties.
The restrictions of random labeling strategies for proteins in general, and antibodies in particular, have admittedly been solved by the development of site-specific labeling protocols. However, the broad use of such protocols is still limited due to the elaborateness of these methods, their suboptimal sensitivity or interference with Fc domain functions. For example, site-specific labeling of IgGs is possible by targeting their N297-linked oligosaccharide chains.Citation3 Due to the importance of N297 glycosylation for FcγR-binding,Citation4 such labeling strategies may affect antibody-FcγR interactions. Site-specific labeling of antibodies has been furthermore achieved by genetic incorporation of unnatural amino acids and subsequent labeling by click-chemistry.Citation5 The method of genetic code expansion for the introduction of unnatural amino acids, however, is not a common standard procedure in higher cells, and there is still need for purification and labeling. The use of antibody fusion proteins with protein domains, such as the SNAP-tag, enabling covalent modification with fluorescent small molecules is less elaborate, but still requires a secondary labeling step.Citation6
Recently, the fluorescent protein citrine has been identified as a versatile antibody labeling domain that may not only be easily incorporated by genetic engineering, but which is also immediately active.Citation7 The comparable low fluorescent activity of citrine (and other fluorescent proteins), however, may limit its use for low expressed antigens. We therefore evaluated the feasibility of antibody labeling by genetic engineering with the luciferase of Gaussia princeps (GpL) as a highly traceable and easily quantifiable labelCitation8 and show here that genetic fusion of GpL to the C-terminus of the antibody light chain has neither a major effect on antigen and Fc-gamma receptor (FcγR)-binding nor on the oligomerization- or FcγR binding-activatable agonistic activity of the antibody irrespective of the isotype. Taken together, we demonstrated that antibody GpL fusion proteins are easily manageable and versatile tools for the functional and kinetic analysis of cell surface antigen-antibody interactions.
Results
Design, generation and production of gaussia princeps luciferase (GpL) fusion proteins of the Fn14-specific IgG1 18D1
We recently demonstrated that genetic fusion of the luciferase from Gaussia princeps (GpL) allows the generation of highly traceable scFv- and tumor necrosis factor superfamily (TNFSF) ligand fusion proteins without disturbing the molecular assembly properties of these molecules and without inference with antigen and receptor binding.Citation9-15 Accordingly, various scFv-GpL and GpL-TNFSF ligand fusion proteins proved to be highly suitable for cellular binding studies and tracer applications. We therefore evaluated whether GpL-tagging is also applicable and useful for conventional antibodies. Initially, we generated different GpL fusion protein formats of the Fn14-specific human IgG1 antibody 18D1 (18D1-IgG)Citation16 to identify the position in the IgG1 molecule that is best suited for linkage of the GpL domain. In one antibody GpL fusion protein format, the GpL domain was linked to the C-terminus of the heavy chain of 18D1 (GpL(CT-HC)-18D1-IgG1), while in 2 other formats the GpL domain has been linked to the N-terminus (GpL(NT-LC)-18D1-IgG1) or C-terminus (GpL(CT-LC)-18D1-IgG1) of the 18D1 light chain (). For gentle purification, we also introduced Flag tags into the various 18D1 constructs. 18D1 and all GpL-18D1-IgG1 variants were transiently expressed in HEK293 cells and secreted with good and comparable efficacy into the cell culture media (). We obtained homogenous and pure non-aggregated preparations of all 18D1-IgG1 variants by affinity chromatography on anti-Flag agarose at physiologic pH ().
Figure 1. Expression and purification of Fn14-specific 18D1-IgG1 antibody GpL fusion proteins. (A) Schemes of the various GpL-18D1-IgG1 fusion protein variants evaluated in this study. (B) The indicated anti-Flag agarose affinity purified GpL-18D1-IgG1 fusion proteins (200 ng) and a marker protein mixture containing 67 ng phosphorylase b, 83 ng albumin, 147 ng ovalbumin, 83 ng carbonic anhydrase, 80 ng trypsin inhibitor and 116 ng α-lactalbumin were resolved by SDS-PAGE and then visualized by silver staining. (C) The various GpL-18D1-IgG1 fusion proteins were analyzed by gel filtration using an aqueous SEC1 column and the following molecular weight marker proteins: bovine thyroglobulin (670 kDa), human IgA (300 kDa), human IgG (150 kDa), ovalbumin (44 kDa) and myoglobin (17 kDa).
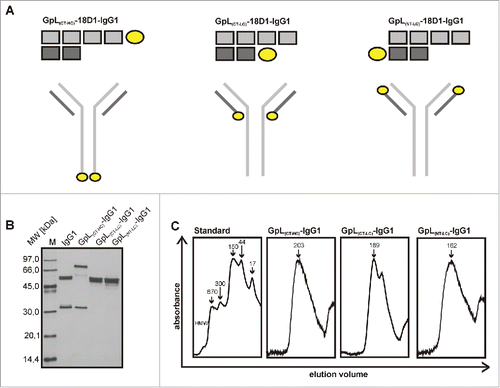
Table 1. Productivity of 18D1-IgG1 variants.
Affinities of GpL-18D1-IgG1 fusion proteins to cell-expressed Fn14 and Fcγ-receptors
To determine the antigen binding properties of the GpL-18D1-IgG1 fusion proteins, we performed cellular equilibrium binding studies with HT1080 cells expressing endogenous Fn14 and a Fn14 knockout variant of this cell line generated using the CRISPR/Cas9 technology (). Pairwise incubation of the Fn14-positive and –negative HT1080 variant revealed Fn14-specific binding of all 3 GpL-18D1-IgG1 variants over a wide range of concentrations (range 0 to 160 nM; ). The calculation of the KD values revealed comparable affinities of 15 and 17 nM for GpL(CT-HC)-18D1-IgG1 and GpL(CT-LC)-18D1-IgG1) and a significantly lower affinity of 74 nM for the GpL(NT-LC)-18D1-IgG1) variant (, ).
Figure 2. Affinity of different GpL-18D1-IgG1 fusion proteins for Fn14. (A) HT1080 and HT1080-Fn14-KO cells were analyzed by FACS with respect to Fn14 expression. (B) HT1080 and HT1080-Fn14-KO cells were seeded in the upper and lower half of a 24-well tissue culture plate. The next day, wells containing the 2 cell types were pairwise incubated (1.5 h, 37°C) with the indicated concentrations of the various GpL-18D1-IgG1 fusion proteins. After removal of the unbound GpL-18D1-IgG1 fusion protein molecules, cell-associated luciferase activity was measured by help of the Gaussia Luciferase Assay Kit. Specific binding values for the various GpL-18D1-IgG1 fusion proteins were calculated by subtracting the non-specific binding values derived of the HT1080-Fn14-KO cells from the Fn14+ parental HT1080 cells. KD values were obtained using the “nonlinear regression to a one-site specific binding curve” function of the GraphPad Prism5 software. Shown is one representative experiment for each fusion protein. (C) Determination of the KD values of the parental Fn14-specific IgG1 antibody 18D1 by heterologous competition binding experiments. The indicated concentrations of 18D1 were mixed with GpL(CT-HC)-18D1-IgG1 (250 ng/ml), GpL(CT-LC)-18D1-IgG1 (100 ng/ml) and GpL(NT-LC)-18D1-IgG1 (250 ng/ml). HT1080 cells were then incubated with these mixtures for 1.5 h at 37 °C. After removal of the unbound antibody fusion proteins, cell-associated luciferase activity was again determined with the Gaussia Luciferase Assay Kit. The binding data obtained were fitted with the “nonlinear regression to a one-site competitive binding curve” function of the GraphPad Prism5 software to calculate the Ki value of 18D1. Shown is one representative experiment for each GpL-18D1-IgG1 fusion protein. Average values and statistics of all experiments are summarized in . RLU, relative light units. (D) Concentration-dependent specific binding of GpL(CT-LC)-18D1-IgG1 to HT1080 cells was determined as in “B.” Specific binding values were normalized against the maximal binding (Bmax) value obtained by nonlinear regression to a one-site specific binding curve. Specific binding values were furthermore converted into the number of occupied Fn14 molecules per cell. (E) Concentration-dependent specific binding of GpL(CT-LC)-18D1-IgG1 and citrine(CT-LC)-18D1-IgG1 to HT1080 cells was determined as in “B.” Specific binding values averaged from 3 independent experiments are shown as fold over background. (F) Equilibrium binding studies with 3 fully independent batches of GpL(CT-LC)-18D1-IgG1 were repeatedly (n = 6) performed as described in “B.” n.s., no significant difference.
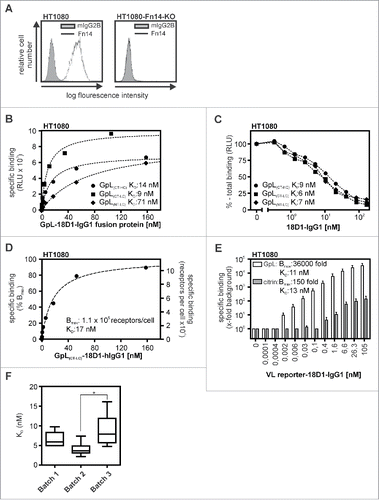
Table 2. Affinity of 18D1-IgG1 variants for cell expressed Fn14.
In competition binding experiments in which the parental non-modified 18D1 antibody was used to inhibit the binding of the GpL-18D1-IgG1 fusion variants to Fn14, we obtained an average Ki value ( = KD value of competitor) of 5 nM, which was not significantly different form the KD values of GpL(CT-HC)-18D1-IgG1 and GpL(CT-LC)-18D1-IgG1 determined in the equilibrium binding studies (, ). Thus, N-terminal positioning of the GpL domain to the variable antibody domain may affect antigen binding, while GpL tagging of the C-terminus of the heavy or light chain has no or only a very moderate effect on antigen affinity. As expected in view of the very high specific bioluminescence activity of Gaussia princeps luciferase,Citation8 the equilibrium binding studies revealed an exquisite sensitivity. The Bmax values obtained with GpL(CT-LC)-18D1-IgG1 in cellular binding studies with ∼200.000 HT1080 cells per sample were ∼30.000-fold over background (). Thus, there was already specific binding with GpL(CT-LC)-18D1-IgG1 at a low concentration of 0.002 nM where only 0.025% of the in average 1.1×105 Fn14 molecules per HT1080 cell are occupied ().
A recent study demonstrated the usefulness of the fluorescent protein citrine as a genetic antibody label for various applications.Citation7 We therefore comparatively analyzed GpL(CT-LC)-18D1-IgG1 with a 18D1 variant in which we replaced the GpL domain by citrine. The resulting citrine(CT-LC)-18D1-IgG1 antibody fusion protein was also suitable in cellular binding studies (). However, while significant binding was already observed with 0.002 nM of GpL(CT-LC)-18D1-IgG1, specific binding of citrine(CT-LC)-18D1-IgG1 started to become detectable only at 0.4 nM (). Accordingly, maximum binding of GpL(CT-LC)-18D1-IgG1 was 36000 fold over background whereas maximum binding of citrine(CT-LC)-18D1-IgG1 was only 150 fold over background (). To get an estimate of the lot-to-lot variation of the functionality of GpL-18D1-IgG1 fusion proteins, 3 batches of independently produced and purified GpL(CT-LC)-18D1-IgG1 were comparatively analyzed in equilibrium binding studies. There were either no significant (batch 1 versus batch 2 and batch 3) or only a minor, ∼factor 2, differences (batch 2 vs. batch 3) between the averaged KD values obtained with these batches (), suggesting an excellent lot-to-lot reproducibility of the functionality of this type of antibody fusion proteins.
Next, we evaluated the various GpL-18D1-IgG1 variants with respect to their binding to FcγRs. For this, we transiently transfected HEK293 cells with human expression plasmids encoding CD16, CD32A, CD32B and CD64 () and used these transfectants to perform equilibrium binding studies at 37°C. Mock transfected cells served to determine the non-specific binding. For all FcγRs with the exception of CD64, there were no significant differences in the affinity of the various GpL-tagged 18D1-IgG1 fusion proteins (, ). Moreover, the KD values obtained were all in the range of the KD values that have been published for IgG1- FcγRs interactions (for review, see ref. Citation17). With respect to CD64 binding, there was no significant difference between the 2 18D1-IgG1 variants with the GpL domain at the C- or N-terminus of the light chain, while GpL(CT-HC)-18D1-IgG1 showed significantly reduced affinity (). The affinities of GpL(NT-LC)-18D1-IgG1 and GpL(CT-LC)-18D1-IgG1) for CD64 were not significantly different from the affinity for CD64 of the parental 18D1 antibody, which was determined by homologous competition experiments (, ). Thus, tagging of the N- or C-terminus of the IgG1 light chain with GpL has no major effect on FcγR binding, while C-terminal tagging of the heavy chain affects CD64 binding. In sum, our binding studies suggested that genetic fusion of the GpL domain to the C-terminus of the 18D1-IgG1 light chain has no major effect on antigen and FcγR binding, and thus identifies this localization as a favorable labeling position.
Figure 3. Affinity of different GpL-18D1-IgG1 fusion proteins for FcγRs. (A) FACS analysis of HEK293 cells transiently transfected with expression plasmids encoding CD16, CD32A, CD32B and CD64. (B) HEK293 cells transiently expressing the indicated FcγRs and mock transfected control HEK293 cells were aliquoted and incubated with increasing concentrations of GpL(CT-HC)-18D1-IgG1, GpL(CT-LC)-18D1-IgG1 and GpL(NT-LC)-18D1-IgG1 (1.5 h, 37°C). After removal of the unbound GpL-18D1-IgG1 molecules cell-associated luciferase activity was determined and specific binding values were calculated by correcting the total binding values obtained from the FcγR transfectants for the unspecific binding values obtained from the mock transfected cells. The KD values were calculated with the “nonlinear regression to a one-site specific binding curve” function of the GraphPad Prism5 software. One representative binding experiment is shown for each of the various interactions. (C) Analysis of the interaction of 18D1 with CD64 by heterologous competition binding experiments with 7.5 ng/ml GpL(CT-HC)-18D1-IgG1, GpL(CT-LC)-18D1-IgG1 and GpL(NT-LC)-18D1-IgG1. Average values and statistics of all experiments are summarized in . RLU, relative light units.
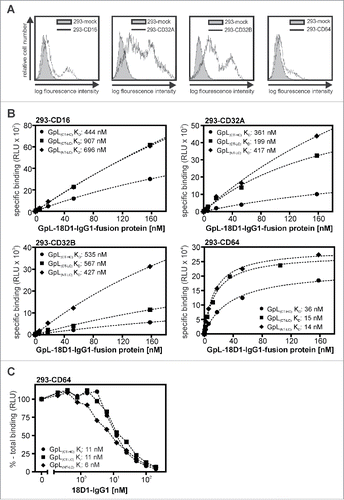
Table 3. Affinity of 18D1-IgG1 variants for cell-expressed FcγRs.
GpL tagging does not interfere with the oligomerization- and FcγR binding-dependent agonistic activity of 18D1-IgG1
Several antibodies that target members of the TNFRSF are currently under preclinical and clinical investigation.Citation18 For several of these antibodies, the IgG1 isoform acquires strong agonistic activity after oligomerization or binding to FcγRs.Citation17, 19-21 The agonism-creating effects of oligomerization and FcγR binding also apply for antagonistic TNFRSF receptor-specific antibodies, and the nature of FcγR type (activating, inhibitory) plays no major role.Citation17, 19-21 The fact that oligomerization or binding to FcγRs converts anti-TNFRSF receptor IgG1 antibodies to agonists presumably reflects the special activation requirements of, at least, some TNFRSF receptors, including CD40, CD95, TRAILR2 and Fn14. For these TNFRSF receptors, there is evidence that binding of 3 receptor molecules by their trimeric TNFSF ligand molecules alone is not sufficient for robust receptor activation and that, in addition to this, secondary aggregation of the initially formed TNFSF ligand-TNFRSF receptor complexes is necessary for a full and strong receptor response.Citation17 Secondary clustering of antibody-bound TNFRSF receptor dimers might be a similar crucial intermediate step in antibody-mediated receptor activation and might be promoted by antibody oligomerizing reagents (e.g., protein G, anti-IgG antibodies) or FcγR-mediated anchoring of the antibody-TNFRSF receptor complexes to the plasma membrane. Thus, not only the antigen and FcγR binding properties could be of relevance for the clinical effects of a TNFRSF receptor-specific IgG1 antibody, but also its ability to form signaling competent supramolecular clusters after oligomerization or FcγR anchoring.
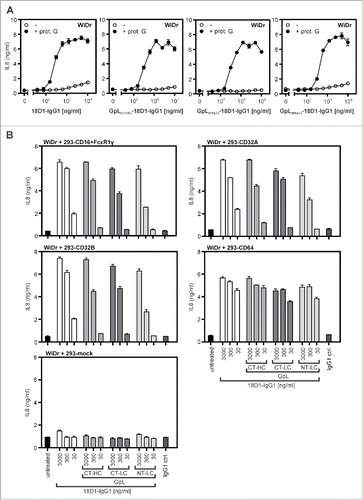
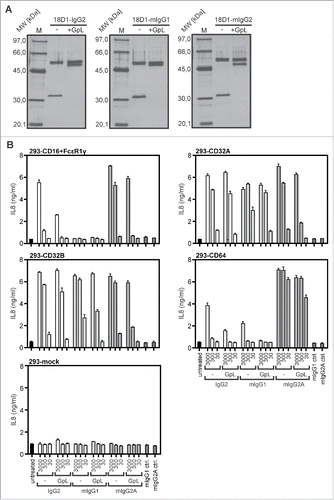
We therefore evaluated whether GpL tagging has an effect on the oligomerization- and FcγR-dependent agonistic activity of 18D1. As shown previously,Citation16 non-oligomerized 18D1-IgG1 largely failed to trigger interleukin (IL)8 production in WiDr cells up to concentrations of 10 µg/ml, while it triggered strong IL8 production already at concentrations below 1 µg/ml in the presence of protein G (). In accordance with the fact that the GpL-18D1-IgG1 fusion proteins do not self-aggregate (), there was also no evidence for these 18D1 variants for an enhanced protein G-independent ability to trigger IL8 production (). Similar to the conventional 18D1-IgG1 variant, all GpL-18D1-IgG1 fusion proteins triggered IL8 production after protein G-treatment. The maximum response and the dose response of the protein G-oligomerized GpL-18D1-IgG1 antibodies were similar to those of their conventional counterpart (). To evaluate the potential of the various 18D1 variants to activate Fn14 upon anchoring to FcγRs, the Fn14-stimulating effect of the antibodies was assayed in co-cultures of WiDr cells and HEK293 cells transiently transfected with expression constructs encoding CD16, CD32A, CD32B and CD64. In cocultures with mock transfected HEK293 cells, none of the 18D1-IgG1 variants showed relevant IL8 induction (). In accordance with the comparable low endogenous Fn14 expression of HEK293 cells and the poor ability of this cell line to produce IL8, the 18D1 variants showed also no major IL8 induction in the FcγR expressing transfectants alone. In all cocultures of WiDr cells and FcγR-expressing HEK293 transfectants, however, the 18D1-IgG1 variants triggered strong stimulation of IL8 production (). All 3 GpL-18D1-IgG1 variants induced IL8 production with efficiency comparable with the conventional 18D1-IgG1 antibody.
Figure 4. Agonistic activity of GpL-18D1-IgG1 fusion proteins upon oligomerization with protein G and FcγR binding. (A) WiDr cells were stimulated in 96-well plates in triplicates with the indicated concentrations of the various GpL-18D1-IgG1 fusion proteins and 18D1 in the presence and absence of 1 µg/ml protein G. The following day, supernatants were collected and analyzed with respect to IL8 production by ELISA. (B) HEK293 cells were transiently transfected with expression vectors encoding CD16, CD32A CD32B, and CD64 or empty vector. The following day, transfectants were mixed with the indicated concentrations of the various 18D1 variants and after 30 min aliquots of 1.7×104 transfected HEK293 cells per well were added to WiDr cells (2 × 104 / well) grown overnight in 96-well plates. Next day, the IL8 content of the supernatants was determined by ELISA.
Our binding and functional studies with GpL(CT-HC)-18D1-IgG1, GpL(NT-LC)-18D1-IgG1 and GpL(CT-LC)-18D1-IgG1 suggest that, especially in the latter variant, the GpL domain has no or only a very minor effect on the properties of the parental conventional antibody. Thus, tagging the light chain C-terminally with a GpL domain appears perfectly suited to generate highly traceable GpL-18D1-IgG1 antibody fusion proteins for in vitro analysis.
Characterization of human and murine IgG1 and IgG2 variants of GpL-18D1 fusion proteins
Besides human IgG1, murine IgG1 (mIgG1), murine IgG2a (mIgG2a) and human IgG2 (IgG2) are additional dimeric antibody isotypes that are used for TNFRSF receptor-targeting in preclinical or clinical studies. We therefore investigated whether C-terminal light chain tagging with a GpL domain is similarly suitable for the characterization of these antibody isotypes. 18D1 variants of human IgG2, mIgG1 and mIgG2a with a light chain having a C-terminal GpL domain (GpL(CT-LC)-18D1-IgG2, GpL(CT-LC)-18D1-mIgG1, GpL(CT-LC)-18D1-mIgG2a) were generated, produced and purified as described above for GpL(CT-LC)-18D1-IgG1 (). With the exception of GpL(CT-LC)-18D1-mIgG1, which showed a slightly but significantly reduced affinity for Fn14, the affinities for Fn14 of the other GpL(CT-LC)-IgG versions of 18D1 were not significantly different (). Moreover, all the new dimeric 18D1 variants displayed no agonistic effect up to concentrations of 3 µg/ml (). As before GpL(CT-LC)-18D1-IgG1 and 18D1-IgG1, all these variants basically converted into potent agonists upon anchoring to FcγRs, although there was somewhat reduced activity with some GpL-variant-FcγR pairings (). Thus, the need of FcγR anchoring for the agonistic activity of 18D1 appears not to be a feature of the human IgG1 isotype, but seems to reflect a general requirement related to the dimeric nature of IgG1 and IgG2 antibodies.
Figure 5. Expression and purification of human and murine GpL(CT-LC)-IgG variants of 18D1. (A, B) Purified GpL(CT-LC)-18D1-IgG2, GpL(CT-LC)-18D1-mIgG1 and GpL(CT-LC)-18D1-mIgG2a (200 ng) were analyzed by SDS-PAGE (A) and gel filtration (B). (C) Equilibrium binding studies with the various novel GpL(CT-LC)-18D1-IgG variants and HT1080 and HT1080-Fn14-KO cells were performed as in to determine their affinity for cell expressed Fn14. Shown is one representative experiment for each fusion protein. (D) Determination of the KD values of 18D1-IgG2, 18D1-mIgG1 and 18D1-mIgG2a by competition binding experiments with 100 ng/ml of GpL(CT-LC)-18D1-IgG2, GpL(CT-LC)-18D1-mIgG1 and GpL(CT-LC)-18D1-mIgG2a.
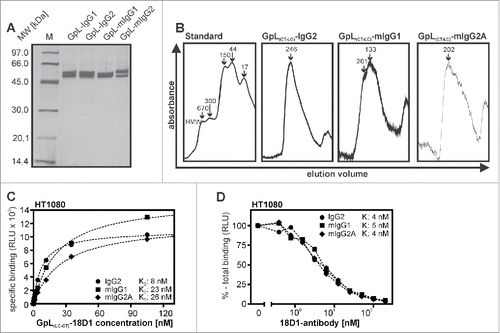
Table 4. Affinity of human and murine 18D1-IgG variants for cell expressed Fn14.
Figure 6. Agonistic activity of human and murine GpL(CT-LC)-18D1-IgG variants upon binding to FcγRs. (A) The indicated purified antibodies and antibody GpL fusion proteins (200 ng) and a marker protein mixture containing 67 ng phosphorylase b, 83 ng albumin, 147 ng ovalbumin, 83 ng carbonic anhydrase, 80 ng trypsin inhibitor and 116 ng α-lactalbumin were evaluated by SDS-PAGE analysis and silver staining. (B) Cocultures of WiDr cells and HEK293 cells transiently transfected with the indicated FcγRs were stimulated overnight in triplicates with 30, 300 and 3000 ng/ml of GpL(CT-LC)-18D1-mIgG1, GpL(CT-LC)-18D1-IgG2, GpL(CT-LC)-18D1-mIgG2a and their GpL-less counterparts. IL8 production was determined by ELISA.
Last but not least, we performed a systematic evaluation of the binding of GpL(CT-LC)-18D1-IgG2, GpL(CT-LC)-18D1-mIgG1 and GpL(CT-LC)-18D1-mIgG2a to human FcγRs. In accordance with the poor KD values that has been reported for the interaction of human IgG2 to CD16 and CD64, we observed no specific binding with GpL(CT-LC)-18D1-IgG2 (). Noteworthy, we obtained in our cellular binding studies medium affinities of 283 and 108 nM for the interaction of GpL(CT-LC)-18D1-IgG2 with CD32A and CD32B, respectively, which are much better than published affinities of IgG2-CD32A/B interactions derived from cell-free experiments ().
Table 5. Affinity of human and murine 18D1-IgG variants for cell expressed FcγRs.
Characterization of GpL fusion proteins targeting CD95, CD40 and LTβR
To clarify whether C-terminal light chain tagging with the GpL domain is straightforwardly transferable to antibodies recognizing cell surface antigens different from Fn14, we generated
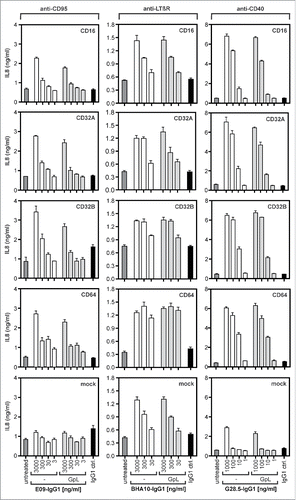
GpL(CT-LC)-IgG1 variants of the CD40-specific antibody G28.5, the CD95-specific antibody E09 and the LTβR-specific antibody BHA10.Citation22, 23, 24 To determine the binding affinities of GpL(CT-LC)-G28.5-IgG1, GpL(CT-LC)-E09-IgG1, GpL(CT-LC)-BHA10-IgG1 and their corresponding conventional human IgG1 variants, we performed again equilibrium binding assays and homologous competition studies. KD values of 5 nM, 4 nM and 2 nM were obtained for GpL(CT-LC)-E09-IgG1, GpL(CT-LC)-G28.5-IgG1 and GpL(CT-LC)-BHA10-IgG1 in equilibrium binding experiments (, ). In competition experiments with varying concentrations of the conventional antibody variants and a fixed concentration of their GpL-tagged counterparts, IC50 values were determined. Based on these values, affinities of the conventional antibody variants of 29 nM (E09-IgG1), 21 nM (G28.5-IgG1) and 10 nM (BHA10-IgG1) were calculated for CD95, CD40 and LTβR binding (, ). The minor differences between the KD values of the conventional and GpL-domain tagged antibody variants obtained were significant (). In view of the very low lot-to-lot variations evident from , it is unlikely that these differences are caused by batch variabilities, suggesting that the GpL domain moderately affected antigen binding in these cases. As with 18D1-IgG1, E09-IgG1 and G28.5-IgG1 showed strongly enhanced receptor stimulating-activity after FcγR-binding, while BHA10-IgG1 revealed an agonistic activity independent of the presence of FcγR-expressing cells (). Intriguingly, the receptor-stimulating abilities of these antibodies were not affected by GpL-tagging (). The results obtained with GpL(CT-LC)-G28.5-IgG1, GpL(CT-LC)-E09-IgG1 and GpL(CT-LC)-BHA10-IgG1 suggest that C-terminal light chain tagging with a GpL domain is a general applicable strategy to generate highly traceable antibodies for cellular binding studies, functional analysis and tracer applications.
Figure 7. Antigen binding of E09-IgG1, BHA10-IgG1, G28.5-IgG1 and their GpL(CT-LC) variants. (A) HEK293 cells and CD95 or LTβR expressing HEK293 transfectants and HT1080 cells and HT1080-CD40 transfectants were seeded in the upper and lower half of a 24-well tissue culture plate and were pairwise incubated the next day (1.5 h, 37°C) with the indicated concentrations of GpL(CT-LC)-E09-IgG1, GpL(CT-LC)-BHA10-IgG1 and GpL(CT-LC)-G28.5-IgG1. Finally, cell-associated luciferase activity was measured and specific binding values for the GpL-IgG1 fusion proteins were obtained by subtracting the non-specific binding values derived of the HEK293 or HT1080 cells from the corresponding receptor transfectants. The “nonlinear regression to a one-site specific binding curve” function of GraphPad Prism5 was used to calculate the KD value. Shown is one representative experiment for each antibody. Averaged values and statistics are summarized in . (B) To determine the KD values of E09-IgG1, BHA10-IgG1 and G28.5-IgG1, the indicated concentrations of these antibodies were mixed with a constant amount of the corresponding GpL(CT-LC) variant (GpL(CT-LC)-E09-IgG1: 3 ng/ml, GpL(CT-LC)-BHA10-IgG1: 100 ng/ml, GpL(CT-LC)-G28.5-IgG1: 1.5 ng/ml). These mixtures were then added for 1.5 h at 37°C to the receptor expressing transfectants and after removal of the unbound antibody fusion proteins cell-associated luciferase activity was measured with the Gaussia Luciferase Assay Kit. The obtained binding data and the KD values determined in “A” for the GpL antibody fusion protein variants were used to calculate the KD value of the conventional IgG1 variants with the “nonlinear regression to a one-site competitive binding curve” function of GraphPad Prism5. Shown is one representative experiment for each antibody and averaged values are again summarized in .
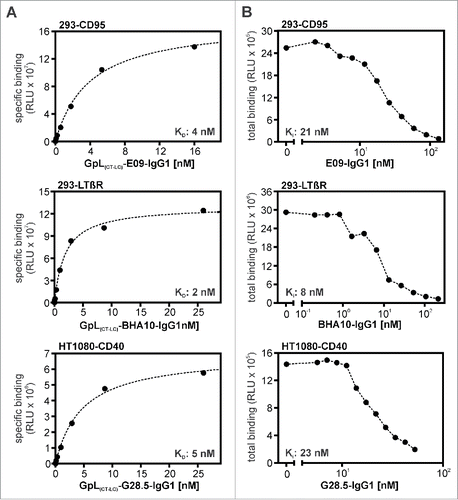
Table 6. Affinity of anti CD95-, anti CD40- und anti LTβR-variants for their antigen.
Figure 8. Agonistic activity of E09-IgG1, BHA10-IgG1, G28.5-IgG1 and their GpL(CT-LC) variants. HT1080-CD40 cells which endogenously express CD95 where cocultured with HEK293 cells transiently transfected with the indicated FcγRs (cotransfection of FcϵR1γ in case of CD16 and CD64) and were stimulated overnight in triplicates with 3000, 300, 30 and 3 ng/ml of E09-IgG1 and 1000, 100, 10 and 1 ng/ml G28.5-IgG1 and the corresponding GpL(CT-LC) antibody variants. In the case of E09-IgG1 20 µM ZVAD and 1 µg/ml CHX were added before stimulation. For coculture with BHA10-IgG1 the same experiment was performed with HT29 cells and 3000, 300 and 30 ng/ml of the antibody. IL8 production was finally measured by ELISA analysis of coculture supernatants.
Discussion
Antibodies raised against cell surface antigens and soluble antigens are of unique relevance for diagnosis and therapy of a variety of disorders. A central step in the development of such antibodies is the characterization of antigen-antibody interaction. Currently, the latter is mainly performed with cell-free techniques such as SPR or isothermal titration calorimetry. These techniques, however, have the limitations briefly discussed in the introduction. In this study, we demonstrated the usefulness of recombinant antibodies fused with the luciferase of Gaussia princeps for cellular binding studies to overcome these limitations. We identified the C-terminus of the antibody light chain as a position for fusing to the GpL domain that does not or only moderately affects antigen binding, and which does not interfere with binding to Fcγ receptors for various human and murine antibody isotypes. Despite the in some cases somewhat reduced affinity of the GpL(CT-LC) antibody fusion proteins, the easily and quite exactly measurable KD values of these fusion proteins allow straightforward and very precise determination of the KD value of the parental “untouched” antibody by competition assays. Interestingly, the linkage of the GpL domain to the antibody light chain had no major negative effect on production and secretion of the antibody-GpL fusion protein (). We showed the reliability not only for our intensively studied Fn14-specific model antibody 18D1, but also for antibodies specific for CD95, LTβR and CD40. Our findings are in good accordance with a study showing that genetic fusion of citrine to the light chain of various antibodies does not affect productivity and antigen affinity of the parental antibodies.Citation7 Together, this suggests that the C-terminus of the antibody light chain is a generally suitable acceptor site for the generation of antibody fusion proteins. Indeed, it turned out that the antibody fusion proteins with C-terminally light chain-tagged GpL not only have normal interaction with their cell surface antigens and Fcγ receptors, but also elicits almost the same oligomerization- and FcγR anchoring-dependent agonistic activity on their target as their conventional counterparts. The fact that the agonistic activity of IgG antibodies targeting Fn14, CD95 and CD40 can be highly dependent on FcγR anchoring is well known and presumably reflects that these TNFRSF receptors require secondary clustering of initially formed ligand-receptor complexes to robustly trigger the full set of receptor associated cellular signaling pathways.Citation17 Noteworthy, the LTβR-specific IgG1 antibody BHA10 and its GpL(CT-LC) variant investigated in our study displayed FcγR binding-independent agonistic activity (). Thus, in sum our functional data suggest that a GpL domain at the C-terminus of the antibody light chain bears a low risk of interference with antibody-specific properties beyond antigen and FcγR recognition. The considerable advantages of the use of antibody GpL fusion proteins are straight forwardly evident. The genetically encoded nature of the label GpL not only circumvent all the technical and reproduction hurdles associated with biochemical labeling methods, but also allows, in the case of cell surface antigens, cellular binding studies avoiding the various problems that can arise from cell-free methods and their need for recombinant antigen and expensive analyzers. The only limitation of the antibody GpL fusion protein format that we have recognized so far is the time required to thoroughly remove unbound molecules from the cell samples. This is performed manually and requires ∼10 sec. for adherent cells and up to one minute with suspension cells. Thus, analyses of antigen-antibody interactions with fast dissociation rates in the range of seconds and below are not straightforwardly possible. Taken together, we showed that antibody GpL fusion proteins are simply available and versatile tools for the characterization of cell surface antigen-antibody with a considerable potential in the early steps of antibody development.
Material and methods
Cell lines and reagents
HEK293, HT29 and HT1080 cells (ATCC, Rockville, MD, USA) were cultivated in RPMI1640 medium (Sigma-Aldrich, Steinheim, Germany) with 10% fetal calf serum (GIBCO, EU. Approved, South America) and WiDr cells were cultivated in DMEM medium (Sigma-Aldrich, Steinheim, Germany) with 10% fetal calf serum. Antibodies used were purchased from eBioscience, San Diego, CA, USA (anti-Fn14-PE, mouse IgG2b, # 12–9018–82; mouse IgG2b-PE, 12–4732–82), Sigma (anti-mouse IgG-PE, polyclonal goat, # P9670 and Santa Cruz Biotechnology, Santa Cruz, CA, USA (anti-CD16-PE, mouse IgG1, # sc-19620; anti-CD64-PE, mouse IgG1, # sc-1184; anti-CD32A/C, mouse IgG1, # sc-373721; anti-CD32B, mouse IgG1, # sc-365864). pCMV-SPORT6 expression vectors encoding FcγRs were from SourceBioscience.
Cloning of antibodies and antibody GpL fusion proteins
DNA stretches encoding the domains of interest flanked by appropriate restriction sites were either synthesized (Geneart, Thermo Fisher Scientific) or generated by PCR. The DNA fragments were sequentially subcloned into the plasmid PS435 (kind gift of Pascal Schneider, University of Lausanne, Epalinges, Switzerland) under removal of the TRAILR2 encoding DNA stretch present in this plasmid. PS435 is based on pCR3 (Invitrogen, Karlsruhe, Germany) and encode an immunoglobulin signal peptide followed by a Flag epitope and TRAILR2.Citation25 Thus, all light and heavy chain-encoding expression constructs contained a N-terminal Flag epitope. The order of the domains in the various constructs is indicated in . The DNA sequences used correspond to amino acids (aa) 18–185 of ac. no. AAG54095.1 (GpL), aa 121–452 of ac. no. AFR78282.1 (CH of human IgG1), aa 1–325 of ac. no. AAN76042.1 (CH of human IgG2), aa 1–106 of ac. no. AAD29610.1 (LC human kappa), aa 143–468 of ac. no. CZF87189.1 (murine IgG1), aa 136–469 of ac. no. S37483 (murine IgG2a) and aa 105–213 of ac. no. ABW24730.1 (LC murine kappa). DNA fragments encoding the variable heavy and light chain sequences of G28.5, E09 and BHA10 were derived from the aa sequences given in GenBank ac. no. AJ853736 (G28.5), the PDB entries 3TJE_L (light chain E09) and 3TJE_H (heavy chain E09) and ref. Citation24 (BHA10).
Production and purification of antibodies and antibody fusion proteins
HEK293 cells were cultivated in 15 cm tissue culture dishes until confluency and medium was changed to 15 ml serum-free RPMI 1640 medium supplemented with penicillin-streptomycin. For each culture dish, 2 ml serum-free RPMI 1640 medium were supplemented with 12 µg of a 1:1 mixture of expression plasmids encoding the light and heavy chain of the antibody of interest. Then 36 µl of a 1 mg/ml polyethylenimine (PEI, Polyscience Inc., Warrington, USA) solution were added dropwise under vortexing, and, after incubation for 15 min at room temperature, the plasmid/PEI solution was added to the HEK293 cells. After overnight cultivation, the serum-free medium was removed and RPMI 1640 medium with 2% FCS and penicillin-streptomycin were added for 4–6 d. Cell culture supernatants were collected and cleared by centrifugation (10 min, 4630 g). The concentration of the antibody or antibody fusion protein was determined by anti-Flag Western blotting and a Flag-tagged standard protein or, where possible, by measuring the activity of the GpL domain.
Antibody and antibody fusion proteins were purified via their internal Flag-tag and anti-Flag affinity chromatography. In brief, an anti-Flag mAb M2 agarose column (1 ml M2 agarose per mg of Flag-tagged antibody/antibody fusion protein) were equilibrated twice with 10 column volumes of TBS. After supplementation with NaCl to 150 mM and immediately before column loading, the cell culture supernatants were again cleared by centrifugation (4630 g, 10 min). Columns were loaded by gravity flow and washed 3 times with 5–10 column volumes of TBS, and finally the Flag-tagged antibody and antibody fusion proteins were eluted with 6–8 aliquots of the column volume of 100 µg/ml Flag peptide (Sigma, Deisenhofen, Germany) in TBS. Fractions were dialyzed against phosphate-buffered saline (PBS), sterile filtered (0.2 µm) and analyzed by SDS-PAGE and silver staining to determine purity and concentration.
SDS-PAGE, silver staining and gel filtration
For verification of the purity and subunit size of the various antibodies and antibody fusion proteins, solutions of samples and molecular weight markers of known concentration and size (Amersham LMW Calibration Kit for SDS Electrophoresis, GE Healthcare) in 2x or 4x Laemmli sample buffer were heated for 5 min at 90°C and subjected to SDS-PAGE on 12.5% (w/v) polyacrylamide gels. The latter were silver stained using the Pierce® Silver Stain Kit (Thermo Scientific, Braunschweig, Germany). Purified antibodies and antibody fusion protein solutions (1000 µl) were subjected to gel filtration on a BioSep-SEC-S3000 (300×7.8) column (Phenomenex, Aschaffenburg, Germany) equilibrated in PBS with a flow rate of 0.5 ml/min to estimate the native weight of the proteins and to obtain information about potential protein aggregation. The gel filtration column was previously calibrated using a marker protein mixture (Column Performance Check Standard, Aqueous SEC 1; Phenomenex, Aschaffenburg, Germany) containing thyroglobulin (670 kDa), human immunoglobulin A (300 kDa) and G (150 kDa), ovalbumin (44 kDa) and myoglobulin (17 kDa).
Binding studies
To determine the affinity of the various antibodies and antibody fusion proteins for Fn14, HT1080 cells expressing endogenous Fn14 and a HT1080 clone in which Fn14 has been disrupted by CRISPR/Cas9 were used. KD values of 18D1-IgG GpL variants were determined by equilibrium binding studies. For this, half of the wells of a 24-well plate were seeded with HT1080 cells and the remaining half of the wells with HT1080-Fn14-KO cells (2×105 cells per well). Next day, the 2 types of cells were pairwise incubated with increasing concentrations of the 18D1-IgG GpL variants for 90 min at 37°C temperature. After removal of unbound proteins by 10 washes (total time ∼5 sec) with ice-cold PBS, cells were harvested in 50 µl RPMI1640 medium with 0.5% FCS using a rubber policeman and transferred to black 96-well plates. GpL activity was determined using the Gaussia luciferase Assay Kit (New England Biolabs GmbH, Frankfurt a.M., Germany) according to the recommendations of the supplier. Light emission was measured within 10 sec after adding the substrate-buffer solution to minimize errors related to the decay of GpL activity (∼ T1/2 = 4 min) using a Lucy 2 or a LUmo Luminometer (both Anthos Labtec Instruments, Krefeld, Germany). Binding values derived of HT1080 cells served as total binding values, while binding values derived of the HT1080-Fn14-KO cells were considered as non-specific binding.
The affinity of GpL(CT-LC)-G28.5-IgG1 for CD40 was similarly determined using HT1080 cells that do not express endogenous CD40 and HT1080-CD40 transfectants.Citation26 The affinities of GpL(CT-LC)-E09-IgG1 and GpL(CT-LC)-BHA10-IgG1 for CD95 and LTβR were furthermore determined using HEK293 cells transiently transfected with empty vector or CD95- and LTβR-encoding expression plasmids. Equilibrium binding studies with citrine(CT-LC)-18D1-IgG1 were essentially performed as described for GpL(CT-LC)-18D1-IgG1 with the exception that cells were finally analyzed in a fluorescence spectrometer (Tecan infinite 200, Männedorf, Switzerland). The antigen affinity of conventional antibodies was determined by using the conventional antibodies as competitive inhibitors of an antibody GpL fusion protein recognizing the same antigen. In brief, increasing concentrations of the conventional antibody of interest (18D1-IgG1, 18D1-mIgG1, 18D1-IgG2, 18D1-mIgG2a, G28.5-IgG1, BHA10-IgG1 or E09-IgG1) were mixed with a constant concentration (in the range of the KD value) of an antibody-GpL fusion protein recognizing the same antigen with known affinity. Antigen-expressing cells (Fn14: HT1080 cells; CD40: HT1080-CD40 transfectants; LTβR: HEK293-LTβR transfectants, CD95: HEK293-CD95 transfectants) were then incubated with these mixtures for 90 min at 37 °C and after removal of the unbound antibody and antibody fusion protein molecules cell-associated luciferase activity was measured.
The determination of the affinity of the various antibody and antibody fusion protein variants for the human FcγRs was performed in principle as described above for the determination of the antigen affinities. Mock transfected HEK293 cells were used to obtain non-specific binding and HEK293 cells transiently transfected with FcγR-encoding expression plasmids were used 1 to 3 d post transfection for measuring of total binding. However, due to the poor plastic adherence of HEK293 cells, incubation with the GpL fusion proteins was done in solution and only 3 washing steps were used to remove unbound proteins. The raw data obtained from the binding studies were analyzed with the GraphPad Prism5 software. Total binding values and corresponding and non-specific binding values derived of equilibrium binding studies were subtracted to obtain specific binding values. KD values were then calculated with the “nonlinear regression to a one-site specific binding curve” function of GraphPad Prism5. Measurements obtained from competition experiments were analyzed with the “nonlinear regression to a one-site competitive binding curve” function of the GraphPad Prism5 software.
Data are reported as mean +/− standard deviation. To evaluate whether affinity differences between 3 or more different antibodies () were significant the KD values derived from independent experiments were analyzed by Bonferroni's test (GraphPad Prism5). In cases where affinity differences between 2 different antibodies were evaluated (), the unpaired t-test was used.
FACS analysis
To evaluate cell surface expression of Fn14 and the various FcγRs, HT1080 cells or HEK293 transfectants were harvested and resuspended in PBS. Cells (0.3 – 2 × 106 in 50 µl) were incubated at 4°C for 0.5 h with primary antibodies of the specificity of interest or appropriate isotype control antibodies at the dilution recommended by the supplier. In cases where non-labeled primary antibodies were used, a PE-labeled secondary antibody was added after one wash in PBS, and, after an additional 30 min at 4 °C, unbound antibodies were removed by 2 washes with PBS. Cells were finally analyzed with a FACSCalibur (BD Biosciences, Heidelberg, Germany).
IL8 ELISA
WiDr cells (2 × 104 / well, 96-well tissue culture plates) were seeded in DMEM containing 10% FCS. The following day, medium was changed to fresh medium supplemented with the different 18D1-antibodies and the HEK293 transfectants of interest or 1 µg/ml protein G. Cells were challenged in triplicates and the next day the IL8 content of the supernatants were evaluated by ELISA (BD Biosciences). For the coculture experiments with E09-IgG1 and G28.5-IgG1, HT1080-CD40 cells (1.5 × 104 / well, 96-well tissue culture plates) were seeded in RPMI1640 medium containing 10% FCS. Next day, medium was changed to fresh medium supplemented again with the antibody variant of interest and the various HEK293 transfectants. The following day, cells were again analyzed in triplicates for the production of IL8 by ELISA (BD Biosciences). In the case of the evaluation of the E09-IgG1 variants, 20 µM of the caspase inhibitor ZVAD were added to prevent cell death induction and 1 µM cycloheximid was added to enhance CD95 signaling. Coculture assays with BHA10-IgG1 were similarly performed with HT29 cells (2 × 104 / well, 96-well tissue culture plates) in RPMI1640 containing 10% FCS.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by Deutsche Forschungsgemeinschaft (grant WA 1025/24–1) and Deutsche Krebshilfe (grant 111703).
References
- Thillaivinayagalingam P, Gommeaux J, McLoughlin M, Collins D, Newcombe AR. Biopharmaceutical production: applications of surface plasmon resonance biosensors. J Chromatogr B Analyt Technol Biomed Life Sci 2010; 878:149-53; PMID:19762290; http://dx.doi.org/10.1016/j.jchromb.2009.08.040
- Velazquez-Campoy A, Ohtaka H, Nezami A, Muzammil S, Freire E. Isothermal titration calorimetry. Curr Prot Cell Biol 2004; Chapter 17:Unit 17.8; PMID:18228446; http://dx.doi.org/10.1002/0471143030.cb1708s23
- Adumeau P, Sharma SK, Brent C, Zeglis BM. Site-specifically labeled immunoconjugates for molecular imaging–part 1: cysteine residues and glycans. Mol Imaging Biol 2016; 18:1-17; PMID:26754790; http://dx.doi.org/10.1007/s11307-015-0919-4
- Subedi GP, Barb AW. The structural role of antibody N-glycosylation in receptor interactions. Structure (London, England : 1993) 2015; 23:1573-83; PMID:26211613; http://dx.doi.org/10.1016/j.str.2015.06.015
- Nikic I, Lemke EA. Genetic code expansion enabled site-specific dual-color protein labeling: superresolution microscopy and beyond. Curr Opin Chem Biol 2015; 28:164-73; PMID:26302384; http://dx.doi.org/10.1016/j.cbpa.2015.07.021
- Cole NB. Site-specific protein labeling with SNAP-tags. Curr Prot Protein Sci 2013; 73: Unit 30.1; PMID:24510614; http://dx.doi.org/10.1002/0471140864.ps3001s73
- Haas AK, von Schwerin C, Matscheko D, Brinkmann U. Fluorescent Citrine-IgG fusion proteins produced in mammalian cells. MAbs 2010; 2:648-61; PMID:20724830; http://dx.doi.org/10.4161/mabs.2.6.13179
- Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther 2005; 11:435-43; PMID:15727940; http://dx.doi.org/10.1016/j.ymthe.2004.10.016
- Fick A, Lang I, Schafer V, Seher A, Trebing J, Weisenberger D, Wajant H. Studies of binding of tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) to fibroblast growth factor inducible 14 (Fn14). J Biol Chem 2012; 287:484-95; PMID:22081603; http://dx.doi.org/10.1074/jbc.M111.287656
- Lang I, Fick A, Schafer V, Giner T, Siegmund D, Wajant H. Signaling active CD95 receptor molecules trigger co-translocation of inactive CD95 molecules into lipid rafts. J Biol Chem 2012; 287:24026-42; PMID:22645131; http://dx.doi.org/10.1074/jbc.M111.328211
- Salzmann S, Lang I, Rosenthal A, Schafer V, Weisenberger D, Carmona Arana JA, Trebing J, Siegmund D, Neumann M, Wajant H. TWEAK inhibits TRAF2-mediated CD40 signaling by destabilization of CD40 signaling complexes. J Immunol 2013; 191:2308-18; PMID:23918987; http://dx.doi.org/10.4049/jimmunol.1202899
- El-Mesery M, Trebing J, Schafer V, Weisenberger D, Siegmund D, Wajant H. CD40-directed scFv-TRAIL fusion proteins induce CD40-restricted tumor cell death and activate dendritic cells. Cell Death Dis 2013; 4:e916; PMID:24232092; http://dx.doi.org/10.1038/cddis.2013.402
- Trebing J, El-Mesery M, Schafer V, Weisenberger D, Siegmund D, Silence K, Wajant H. CD70-restricted specific activation of TRAILR1 or TRAILR2 using scFv-targeted TRAIL mutants. Cell Death Dis 2014; 5:e1035; PMID:24481449; http://dx.doi.org/10.1038/cddis.2013.555
- Bittner S, Knoll G, Fullsack S, Kurz M, Wajant H, Ehrenschwender M. Soluble TL1A is sufficient for activation of death receptor 3. Febs j 2016; 283:323-36; PMID:26509650; http://dx.doi.org/10.1111/febs.13576
- Lang I, Fullsack S, Wyzgol A, Fick A, Trebing J, Arana JA, Schäfer V, Weisenberger D, Wajant H. Binding studies of TNF receptor superfamily (TNFRSF) receptors on intact cells. J Biol Chem 2016; 291:5022-37; PMID:26721880; http://dx.doi.org/10.1074/jbc.M115.683946
- Trebing J, Lang I, Chopra M, Salzmann S, Moshir M, Silence K, Riedel SS, Siegmund D, Beilhack A, Otto C, et al. A novel llama antibody targeting Fn14 exhibits anti-metastatic activity in vivo. MAbs 2014; 6:297-308; PMID:24135629; http://dx.doi.org/10.4161/mabs.26709
- Wajant H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ 2015; 22:1727-41; PMID:26292758; http://dx.doi.org/10.1038/cdd.2015.109
- Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012; 119:651-65; PMID:22053109; http://dx.doi.org/10.1182/blood-2011-04-325225
- Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res 2013; 19:1035-43; PMID:23460534; http://dx.doi.org/10.1158/1078-0432.CCR-12-2064
- DiLillo DJ, Ravetch JV. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol Res 2015; 3:704-13; PMID:26138698; http://dx.doi.org/10.1158/2326-6066.CIR-15-0120
- Nimmerjahn F, Gordan S, Lux A. FcgammaR dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends Immunol 2015; 36:325-36; PMID:25981969; http://dx.doi.org/10.1016/j.it.2015.04.005
- Clark EA, Yip TC, Ledbetter JA, Yukawa H, Kikutani H, Kishimoto T, Ng MH. CDw40 and BLCa-specific monoclonal antibodies detect two distinct molecules which transmit progression signals to human B lymphocytes. Eur J Immunol 1988; 18:451-7; PMID:2451615; http://dx.doi.org/10.1002/eji.1830180320
- Chodorge M, Zuger S, Stirnimann C, Briand C, Jermutus L, Grutter MG, Minter RR. A series of Fas receptor agonist antibodies that demonstrate an inverse correlation between affinity and potency. Cell Death Differ 2012; 19:1187-95; PMID:22261618; http://dx.doi.org/10.1038/cdd.2011.208
- Michaelson JS, Demarest SJ, Miller B, Amatucci A, Snyder WB, Wu X, Huang F, Phan S, Gao S, Doern A, et al. Anti-tumor activity of stability-engineered IgG-like bispecific antibodies targeting TRAIL-R2 and LTbetaR. MAbs 2009; 1:128-41; PMID:20061822; http://dx.doi.org/10.4161/mabs.1.2.7631
- Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, Holler N, Tschopp J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 1997; 7:831-6; PMID:9430228; http://dx.doi.org/10.1016/S1074-7613(00)80401-X
- Wyzgol A, Muller N, Fick A, Munkel S, Grigoleit GU, Pfizenmaier K, Wajant H. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J Immunol 2009; 183:1851-61; PMID:19596991; http://dx.doi.org/10.4049/jimmunol.0802597