
ABSTRACT
Standardized and biologically relevant potency assays are required by the regulatory authorities for the characterization and quality control of therapeutic antibodies. As critical mechanisms of action (MoA) of antibodies, the antibody-dependent cell-meditated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) must be characterized by appropriate potency assays. The current reference method for measuring cytotoxicity is the 51Cr-release method. However, radioactivity handling is difficult to implement in an industrial context because of environmental and operator protection constraints. Alternative non-radioactive methods suffer from poor validation performances and surrogate assays that measure FcγR-dependent functions do not comply with the regulatory requirement of biological relevance. Starting from these observations, we developed a non-radioactive luminescent method that is specific for target cell cytolysis. In adherent and non-adherent target cell models, the ADCC (using standardized effector cells) or CDC activities of rituximab, trastuzumab and adalimumab were compared in parallel using the 51Cr or luminescent methods. We demonstrated that the latter method is highly sensitive, with validation performances similar or better than the 51Cr method. This method also detected apoptosis following induction by a chemical agent or exposure to ultraviolet light. Moreover, it is more accurate, precise and specific than the concurrent non-radioactive calcein- and TR-FRET-based methods. The method is easy to use, versatile, standardized, biologically relevant and cost effective for measuring cytotoxicity. It is an ideal candidate for developing regulatory-compliant cytotoxicity assays for the characterization of the ADCC, CDC or apoptosis activities from the early stages of development to lot release.
Abbreviations
| Ab | = | antibody |
| ADCC | = | antibody-dependent cell-meditated cytotoxicity |
| CDC | = | complement-dependent cytotoxicity |
| CI | = | confidence interval |
| CV | = | coefficient of variation |
| E:T | = | effector:target |
| FBS | = | fetal bovine serum |
| RLU | = | relative luminescence units |
| S/B | = | signal/background |
| SD | = | standard deviation |
| TR-FRET | = | time-resolved fluorescence resonance energy transfer |
| USP | = | United States Pharmacopeial Convention |
Introduction
International regulatory authorities, such as the Food and Drug Administration or European Medicines Agency, require the inclusion of a wide panel of characterization approaches in the quality control process associated with therapeutic antibody (Ab) production.Citation1,2 In addition to the physicochemical (e.g., sequence) and immunological (e.g., affinity, cross-reactivity) properties or purity and contaminant quantification, the biological activity must be characterized by using the appropriate in vitro potency assays. This important role of potency assays has been further strengthened by the expansion of the biosimilar market because biological activity tends to be considered a key factor in demonstrating biosimilarity.Citation3-5 According to the regulatory definition, the potency represents “the quantitative measure of biological activity based on an attribute of the product which is linked to the relevant biological properties.”Citation1 Potency assays are used to characterize the product and monitor lot-to-lot consistency and stability;Citation2 thus, highly precise, accurate and robust assays must be developed to ensure the therapeutic product reliability. Similar to all characterization and quality control purposed assays, potency assays must be validated according to very strict guidelines, such as the International Conference on Harmonization (ICH) Q2 (R1)Citation6 or the United States Pharmacopeial Convention (USP) chapters <1032>, <1033> and <1034>.Citation7
The induction of cytotoxicity against target antigen-expressing cells is a key mechanism of action of several therapeutic Abs, particularly in the case of anti-tumor human IgG1. This cytotoxic activity may be mediated directly through antigen binding, which leads, for example, to apoptosis induction, or to cell killing through immune effector mechanisms that involve the Fc portion of the Ab and result in the complement cascade activation (CDC) or the recruitment of Fc receptor-expressing cytotoxic effector cells (e.g., natural killer cells or macrophages) that mediate target cell lysis via ADCC or phagocytosis.Citation8 Numerous publications highlight the role of the interaction between the Fc portion of IgG1 and Fcγ receptors, and particularly FcγRIIIa (CD16a) in humans, in the functional response to several therapeutic antibodies in vitroCitation9 and in vivo,Citation10-12 assigning a central role to the ADCC in the mechanism of action of these antibodies. Furthermore, the improvement of CD16a binding by glyco- or protein-engineering has yielded Fc-optimized antibodies with higher in vivo efficacy in pre-clinical and clinical studies,Citation13-16 which has resulted, for example, in the recent approval of Gazyvaro® (anti-CD20, obinutuzumab).Citation17
In this context, the availability of a relevant potency assay to measure Ab-induced cytotoxicity and, more specifically ADCC activity, is a key factor in the development of therapeutic antibodies to ensure candidate screening, production optimization and lot-to-lot consistency. Initially reported in the 1960s,Citation18,19 the commonly used 51Cr-release assay (similar to other radionuclide-based assays) has been considered the most sensitive and biologically relevant assay for cytotoxicity. As a result of the relatively low level of 51Cr spontaneous release by the radiolabeled cells and the high sensitivity provided by the radioactive signal, the method is sensitive and provides a good signal/background (S/B) ratio, even in the presence of a limited number of target cells per test (1,500 to 3,000 cells). These characteristics result in good performances in terms of accuracy, precision and robustness, at least for a complex bioassay, such as an ADCC assay. In addition, the underlying mechanism of 51Cr release is fully consistent with the biological phenomenon of the ADCC and is thus compliant with regulatory requirements regarding potency assays. However, taking into consideration the advances in environmental protection and operator safety, the use of radionuclides is increasingly constraining and costly and is nearly impossible to implement in an industrial context.
Several non-radioactive alternatives to the 51Cr-release assay have been described or are commercially available. These methods are based on a direct cell death measurement or an indirect measurement of a surrogate event more or less closely associated with cell death. The direct methods include target cell labeling with non-radioactive molecules, such as calcein or time-resolved fluorescence resonance energy transfer (TR-FRET) probes (e.g., lanthanide chelates), which may be detected following cell death-induced release on a principle similar to the 51Cr-release method. However, these methods are dependent on the activity of intracellular esterases (required to activate the cell-permeable pro-forms of the reporter molecules in the cytoplasm), which results in target cell line-dependent labeling variations. Moreover, both calcein and lanthanides exhibit high to very high levels of spontaneous release,Citation20-22 which result in low sensitivity assays compared with the 51Cr-release assay, despite the higher number of target cells required per assay (classically 5,000 to 15,000). Another group of direct and specific methods for evaluating target cell death in an ADCC assay is based on flow cytometry.Citation23-25 Combining differential labeling of target and effector cells with viability markers, these methods specifically measure target cell death or disappearance. However, they also suffer from 2 classical limitations of flow cytometry, low throughput and relatively high sample-to-sample variations, which result in insufficient precision and robustness for regulatory-compliant use (internal unpublished results).
A third group of methods that directly measure cell death is based on the measurement of ubiquitary and constitutively expressed enzymes or molecules Citation26-28 released during the cytolytic process. Despite good performances in terms of the validation potential for evaluating the death of a homogeneous cell population, these methods are not pertinent when 2 or more cell types are co-cultured in a single well, which is, for example, the case in an ADCC assay. Effector cells that die during the cytolytic process will participate in the global enzymatic release, and it will not be possible to discriminate between the target- and effector-originating signals. A similar reasoning may be applied to impedance-based methods, in which the cell adherence is measured thanks to an impedance-sensitive chip.Citation29,30 In addition to being limited to the sole use of adherent target cells, this method only evaluates the growth or disappearance of cells through their adhesion to the substrate; it does not measure cell death. Overall, there is no fully satisfactory non-radioactive method for measuring target cell death in the context of a regulatory-compliant ADCC assay with good validation performances.
To circumvent these difficulties in validating ADCC assays that measure the actual target cell death, various indirect surrogate methods have been developed. In general, these surrogate assays are based on a measurement of CD16-dependent functions using FcγR-expressing reporter cell lines associated with an appropriate reporter system. For example, Promega markets a Jurkat cell line that is genetically transformed to express CD16 (for an ADCC-like assay) or CD32 (for an Ab-dependent cellular phagocytosis [ADCP]-like assay) and a luciferase reporter gene under the dependence of the FcγR-associated NFAT signaling pathway. In anti-CD20 models, the method exhibited good validation potential.Citation31 Several other methods are based on a similar principle of measuring reporter effector cell activation in response to an Ab to extrapolate its ADCC activity.Citation32,33 Nevertheless, the main drawback of these methods is that they assume that the CD16-dependent response is predictive of the ADCC activity, and there are several reports that provide findings against this assumption.Citation34,35 For example, in 2009, Kawaguchi et al. demonstrated that the physiologic state or the initiation of mechanisms of resistance to the perforin/granzyme B pathway may modify the ADCC sensitivity of target cells.Citation35 These findings are likely model-dependent; however, in the absence of a clear demonstration that the CD16-dependent response and ADCC activity are correlated in a specific model, these surrogate assays, which are based on the activation of effector cells, are not compliant with the rules of regulatory authorities regarding potency assays.
Based on these different observations, we aimed to develop a non-radioactive method with the objective of regulatory compliance. The method should be specific for target cell lysis, easy to use, based on an inexpensive and widely spread readout method and should exhibit similar or better performances than the 51Cr-release assay.
Results
Selection of a reporter molecule
Our initial objective was to develop a method based on a principle similar to the 51Cr-release gold standard method, i.e., measuring an intracytoplasmic molecule that is released in the culture supernatant by the dying target cells. This molecule had to be specifically expressed by the target cells as a free form in the cytoplasm and exhibit release kinetics comparable to 51Cr release; for example, it should reach its maximum 3 to 6 hours following reaction initiation in the case of an ADCC assay. To avoid the use of an extemporaneous chemical labeling (such as calcein or 51Cr), which is a source of potential inter-assay variability, we chose to construct genetically transformed target cells that constitutively express a reporter protein.
Several reporter proteins have been tested, which vary in size and readout technology. Target cell lines were transfected with an expression vector that encoded the tested reporter protein, and the screening parameters for evaluating these molecules included the linearity of the signal, the signal/background (S/B) ratio, and the kinetics of the release. We first tested a 47 kDa chimerical protein, which consisted of 2 tag sequences and was detected with specific antibodies through a lanthanide-based TR-FRET readout. Despite a bright signal following cell lysis with Triton X-100, which resulted in a good S/B ratio (> 15), no protein release was identified in a classical ADCC set up after 4 hours of incubation. Moreover, at least 20 hours were required to identify significant release, which led to highly variable results (data not shown).
We subsequently decided to test the nanoluciferase protein. Nanoluciferase is a recently described Citation36 protein of 19 kDa derived from the catalytic subunit of the deep-sea shrimp Oplophorus gracilirostris. Two target cell models were constructed to stably express nanoluciferase, the non-adherent Raji B cell lymphoma and the adherent SKOV-3 ovarian cancer cell lines. Preliminary data using nanoluciferase-containing cell culture supernatants demonstrated that the signal was linear between ∼3,000 and 1,000,000 relative luminescence units (RLU), which enabled a wide range analysis of 2.5-Log10. We subsequently assessed the efficiency and sensitivity of the method in both cellular models in the presence of decreasing amounts of nanoluciferase-expressing target cells by evaluating both the linearity of the spontaneous and maximum release signals and the resulting S/B ratio in typical ADCC assay conditions. We used 3,000, 1,500, 1,000 and 500 cells per well, with or without lysis with Triton X-100, and the results are shown in . The data initially confirmed the linearity of the signal in the previously defined range and a typical ADCC assay experimental setting. For Raji cells, the maximum signal did not exceed 1.5×105 RLU, and the linear determination coefficients r2 were 0.9731 and 1.0000 for the spontaneous and maximum release signals, respectively (). The resulting S/B ratios were approximately constant, regardless of the amount of cells, which ranged from 7.0 to 9.4 (). For the SKOV-3 cells, the maximum release signal ranged from 2.7×105 to 1.0×106 RLU for 500 and 3,000 cells per well, respectively (). The coefficients r2 were 0.9936 and 0.9949 for the spontaneous and maximum release signals, respectively. The resulting S/B ratios ranged from 14.5 using 500 cells/well to 7.8 using 3,000 cells ().
Figure 1. Characterization of the spontaneous and maximum release signals of nanoluciferase-expressing cells. Varying numbers of nanoluciferase-expressing Raji (A and B) or SKOV-3 (C and D) cells were incubated in triplicates for 4 h at 37°C in RPMI-1640 with 5% FBS that contained an irrelevant monoclonal Ab (trastuzumab for the Raji cells and rituximab for the SKOV-3 cells) and ADCC effector cells (E:T = 10:1), in the presence or absence of Triton X-100 to measure the maximum and spontaneous release signals, respectively, in a typical ADCC experimental setting. The nanoluciferase signal was measured as described in the Materials and Methods section. Left graphs (A and C) represent the measured spontaneous (open circles) or maximum (black squares) signals released by the different amounts of cells (mean ± SD). Linear regressions were performed for each set of data and are shown in the left graphs (dotted line for spontaneous and solid line for maximum release). For each evaluated amount of cells per well, the S/B ratio was calculated as the mean of the maximum signal divided by the mean of the spontaneous release and is shown in the right graphs (B and D).
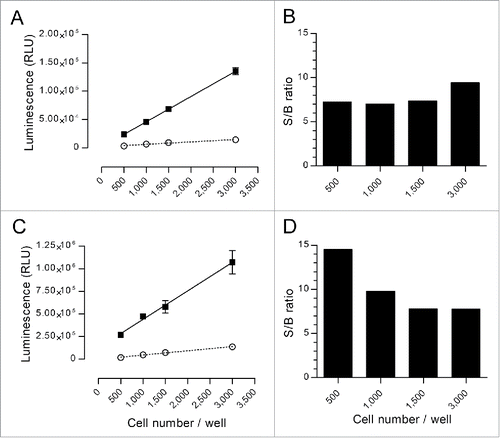
These data demonstrated that very good S/B ratios were achieved with the nanoluciferase method, even with a very low number of target cells. Consistently, very satisfactory spontaneous release percentages were demonstrated with nanoluciferase, thereby reaching 10% and 13% for the Raji and SKOV-3 cells, respectively, regardless of the amount of cells. For comparison, our data exhibited 18% and 23%, respectively, in the same conditions using 51Cr labeling.
Assessment of nanoluciferase release as a consistent marker of cell death in several cytotoxicity models
The next step was to demonstrate that the nanoluciferase release was correlated with actual cell death through a physiologic mechanism. Thus, we induced apoptosis in the nanoluciferase-expressing Raji clone, with increasing doses of staurosporine or increasing durations of ultraviolet (UV) light exposure. After 48 h of culture, the nanoluciferase signal was measured in the supernatants, and the viability of the cells was measured via flow cytometry in the corresponding pellets. The percentages of dead cells were calculated from both methods, and the results are shown in . A very good correlation was found between both methods, with a regression slope approximately equal to 1 (0.97 for both UV and staurosporine induction), regardless of the type of apoptosis induction, i.e., physical or chemical. This finding demonstrated that the nanoluciferase release method was significantly representative of the target cell death and directly proportional to the actual dead cell percentage.
Figure 2. Correlation between cell death and nanoluciferase release. 5.0×104 nanoluciferase-expressing Raji cells/well were cultured for 48 h in RPMI-1640 with 10% FBS [A] in the presence of increasing doses of the apoptosis-inducing drug staurosporine (6,4 nM to 20 µM) or [B] after increasing periods of UV exposure (10 to 60 s). In each well, the percentage of dead cells was calculated by measuring the nanoluciferase signal in the supernatant and via flow cytometry on the cell pellets, as described in the Materials and Methods section. For each tested condition in 2 independent assays, the graphs summarize the percentage of dead cells based on the nanoluciferasedetermination (x-axis) vs. the percentage of dead cells based on the flow cytometry method (y-axis). The dotted line indicates the linear regression calculated from the data. The regression equations were y = 0.97x – 2.75 (r2 = 0.9830) for staurosporine-induction (A) and y = 0.97x – 4.90 (r2 = 0.9595) for UV induction (B).
![Figure 2. Correlation between cell death and nanoluciferase release. 5.0×104 nanoluciferase-expressing Raji cells/well were cultured for 48 h in RPMI-1640 with 10% FBS [A] in the presence of increasing doses of the apoptosis-inducing drug staurosporine (6,4 nM to 20 µM) or [B] after increasing periods of UV exposure (10 to 60 s). In each well, the percentage of dead cells was calculated by measuring the nanoluciferase signal in the supernatant and via flow cytometry on the cell pellets, as described in the Materials and Methods section. For each tested condition in 2 independent assays, the graphs summarize the percentage of dead cells based on the nanoluciferasedetermination (x-axis) vs. the percentage of dead cells based on the flow cytometry method (y-axis). The dotted line indicates the linear regression calculated from the data. The regression equations were y = 0.97x – 2.75 (r2 = 0.9830) for staurosporine-induction (A) and y = 0.97x – 4.90 (r2 = 0.9595) for UV induction (B).](/cms/asset/930fbcb0-6047-4453-bdc8-42653323f1fa/kmab_a_1286435_f0002_b.gif)
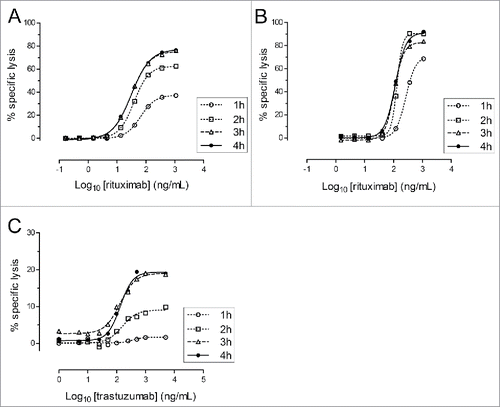
We subsequently assessed the kinetics of the nanoluciferase release in ADCC and CDC assays, particularly in comparison to the 51Cr-release method, which are classically performed during 3 to 4 hours. The ADCC and CDC assays were thus performed using increasing incubation times of 1, 2, 3 or 4 hours using Raji cells ( and ). The SKOV-3 cell line, similar to many carcinoma-derived cell lines, strongly expressed complement resistance factors Citation37 and was thus non-sensitive to the CDC; thus, only the ADCC activity was tested with this model (). For the ADCC assays, the nanoluciferase release was time-dependent, with an activity plateau reached after 3 hours in both models. For the CDC, the maximum lysis was obtained after 2 hours. Overall, these assay durations were completely similar to the classical 51Cr-release based method.
Figure 3. Kinetics of nanoluciferase release in ADCC and CDC assays. Nanoluciferase-expressing non-adherent Raji (A and B) or adherent SKOV-3 (C) cells were used in the ADCC (A and C) or CDC (B) assays performed during 1, 2, 3 or 4 h. At the end of the incubation, supernatants were collected, and the nanoluciferase activity was measured. The graphs indicate the percentage of the resulting specific lysis (mean of triplicates, y-axis) for each Ab concentration (Log10-transformed, x-axis) and the corresponding 4-parameter logistic regression models. The data were obtained from a single experiment, with each condition performed in triplicate.
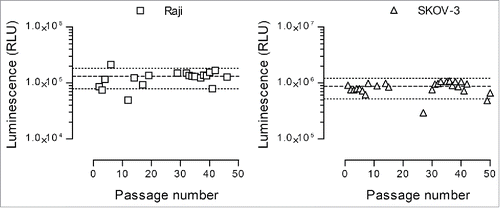
Moreover, an important consideration to ensure the reproducibility of the cytotoxicity experiments was the stability of the nanoluciferase expression. Hence, this stability was assessed with both the Raji and SKOV-3 selected clones during 50 passages, and the results are shown in . Three thousand cells were used for each determination, and the average RLU values (mean ± SD, coefficient of variation [CV] between brackets) along the 50 passages were 125,674 ± 36,505 RLU (29.1%) for the Raji clone (n = 20 determinations) and 842,262 ± 190,569 RLU (22.6%) for the SKOV-3 clone (n = 26 determinations). Taking into account that the experimental process involved a manual counting step (with 20% potential variation), a variability of 40% of the median RLU value was considered acceptable. Consequently, with the exception of few outliers, the stability of the nanoluciferase expression was considered satisfactory.
Figure 4. Nanoluciferase expression stability. The selected nanoluciferase-expressing Raji (left graph) and SKOV-3 (right graph) clones were monitored during 50 passages for nanoluciferase expression. Three thousand cells were lysed with Triton X-100, and the resulting luminescence signal was measured following substrate addition as described in the Materials and Methods section. Each dot represents an individual determination, and the median ± 40% is shown as large and small dotted lines, respectively.
Comparison of nanoluciferase- and 51Cr-release methods in ADCC and CDC assays
The nanoluciferase-release method was then directly compared with the 51Cr-release method in ADCC and CDC assays. To improve the ADCC assay reproducibility, a human CD16-transduced cytotoxic T lymphocyte clone was used following standardized production.Citation38 As a non-adherent target cell model, nanoluciferase-expressing Raji cells were used in both the ADCC and CDC experiments. Because of their resistance to CDC, the SKOV-3 adherent cell model could only be used for the ADCC assay. Thus, as an adherent target cell model for the CDC assay, we selected an internally developed Chinese hamster ovary (CHO) cell line that expressed a low-cleavage mutant form of membrane tumor necrosis factor (TNF) (CHO/TNF, unpublished data), which has also been transformed to stably express nanoluciferase. The average nanoluciferase expression of the CHO/TNF clone was similar to the previously described SKOV-3 clone (824,566 ± 162,105 RLU).
Three independent ADCC or CDC experiments were performed. For each target cell model, nanoluciferase-expressing cells were or were not labeled with 51Cr, and both were run in parallel in the ADCC assay (using rituximab for Raji cells and trastuzumab for SKOV-3 cells) or in the CDC assay (using rituximab for Raji cells and adalimumab for CHO/TNF cells). In each assay, Ab preparations, effector cell suspensions and complement dilutions were shared between the 51Cr-labeled and unlabeled target cells. After 4 h of incubation, the specific lysis was measured using a 51Cr- or nanoluciferase-release readout. The results are shown in , and the model characteristics are summarized in . Each condition was tested in triplicates and average intra-replicate variations were 3.8% and 7.3% for 51Cr- and nanoluciferase-based methods, respectively. For both assays (ADCC and CDC) and both models (adherent and non-adherent target cells), the nanoluciferase-based method provided very similar results to the 51Cr-based method. A deeper analysis of the modeling parameters () indicated nearly identical Emin and Emax values between both methods for these 2 models. The EC50 were also very comparable between both methods; however, a significantly lower value was identified with the nanoluciferase method (p = 0.0093, paired non-parametric Wilcoxon's test, n = 12), with a ∼2-fold factor, with the exception of the CHO/TNF CDC model. An analysis of the EC50 CVs also confirmed the similarity of both methods in terms of the inter-assay variability, with a slight, but not significant (paired t test), advantage for the nanoluciferase-based method, which generally exhibited smaller CVs (7.2% to 22.8% versus 18.6% to 38.4%, respectively).
Figure 5. Direct comparison between 51Cr- and nanoluciferase-based methods. ADCC assays (A and B) were performed using nanoluciferase-expressing Raji cells (A) as a non-adherent target cell model and SKOV-3 cells (B) as an adherent target cell model. The CDC assays (C and D) were performed using nanoluciferase-expressing Raji (C) and CHO/TNF cells (D) as non-adherent and adherent target cell models, respectively. Cells were or were not labeled with 51Cr and were subsequently run in parallel in the ADCC or CDC assays. The percentages of specific lysis (y-axis) obtained in the presence of the indicated Ab concentrations (Log10-transformed, x-axis) are indicated on the graphs, with the 51Cr-release method (open squares) or the nanoluciferase-release method (black circles), together with the calculated unconstrained 4-parameter logistic regression models (dotted and solid lines, respectively). The data were obtained from 3 independent assays (mean ± SD).
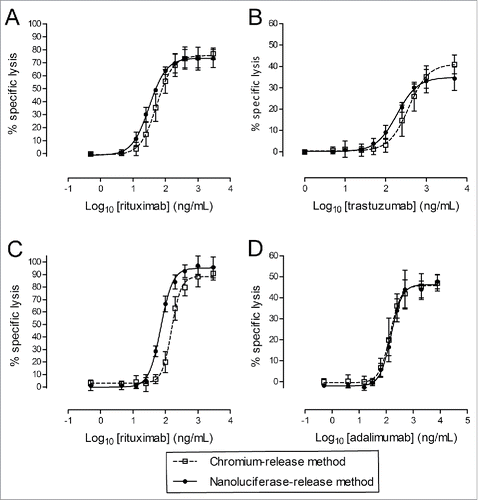
Table 1. Comparison of the characteristic parameters of ADCC and CDC assays using 51Cr and nanoluciferase methods in the unconstrained 4-parameter logistic regression model. Arithmetic mean values and ranges (minimum/maximum) of the EC50 (in ng/mL), Emin and Emax (both in % of specific lysis), as well as the coefficient of variation (CV) of the EC50 are indicated in the table for both the ADCC and CDC assays using 51Cr or nanoluciferase readouts. The data were obtained from 3 independent experiments.
Performance of the nanoluciferase assay compared with the 51Cr-release assay
A cytotoxicity assay compliant with the regulatory guidelines on monoclonal Abs must follow international guidelines regarding data analysis and validation of the performance criteria, such as the accuracy, precision or robustness. Accordingly, we designed a validation procedure based on both the ICH Q2(R1) guidelines Citation6 and the recently published USP chapters <1032>, <1033> and <1034> regarding Bioassay Validation. In addition to the standard Ab dilution ranges described in the previous paragraph and , which is considered the “reference sample”, each reference Ab was diluted or concentrated at 80% or 125% of the nominal concentration to yield under- and over-potent samples (which mimicked batch-to-batch variations from the same mAb). These “test samples” were subsequently run in parallel with the reference sample in both the 51Cr- and nanoluciferase-based ADCC and CDC assays, using nanoluciferase-expressing target cells in both methods. To follow a worst-case approach and obtain a preliminary idea of an assay's robustness, the 3 assays were performed by 2 different operators on 2 different days for each model.
The amount of data available to us was too small to calculate equivalence margins as recommended by the USP equivalence testing approach, which prevented the calculation of a “relative potency” in the strict regulatory meaning. Thus, we based our analysis on the EC50 ratio between the reference and test samples in the unconstrained model, as a surrogate of a strict relative potency calculation. For each test sample, the bias (i.e., the ratio between the measured and expected “potency”) was subsequently calculated. This analysis was performed in each model for the 3 assays in both methods, and the results are summarized in .
Table 2. Summary of EC50 ratios and resulting biases calculated for under- (80%) and over- (125%) potent samples in the different ADCC and CDC models. The ratio between the reference and the under- or over-potent samples was calculated for each assay, and the table indicates the arithmetic mean values and CV. The biases represent the difference between the calculated and expected (0.8 and 1.25 for 80% and 125% samples, respectively) ratios. The table indicates the biases' arithmetic mean values and ranges (minimum/maximum). The data were obtained from 3 independent experiments.
Globally, both methods exhibited a similar potential to potency calculation, despite small differences within each model. For the ADCC assay with Raji cells, the EC50 ratios and biases were comparable between the 51Cr and nanoluciferase methods (∼5%); however, a higher CV was identified with nanoluciferase (26% and 21% for the 80% and 125% test samples, respectively, compared with 14% and 9% for 51Cr). In the adherent cell ADCC model (SKOV-3), 51Cr had slightly higher CVs (15.7% and 16.1% for both test samples, respectively, compared with 10.7% and 14.9% for nanoluciferase), a lower bias for the 80% sample (3.6% vs. −13.4%, respectively) and a higher bias for the 125% sample (23.4% vs. 7.7%, respectively). The same types of small variations were identified in the CDC models, in which 51Cr globally exhibited slightly better performances in the non-adherent cell model, but slightly worse performances in the adherent cell model. Overall, both methods appeared to provide very similar results, which confirmed the comparability of the release mechanisms identified in the previous figures between 51Cr and nanoluciferase.
Comparison of the nanoluciferase assay to the other non-radioactive methods calcein and TR-FRET
We finally compared the nanoluciferase assay to 2 other non-radioactive methods already available for cytotoxicity measurement, the calcein release method and the DELFIA® (from Perkin Elmer) TR-FRET method. The principle of these methods is to label the target cells with acetoxymethyl (AM) pro-forms of calcein or of the fluorescence enhancer terpyridine-6,6″-dicarboxylate (TDA). The AM moiety is then cleaved by non-specific intracellular acetyl esterases, decreasing the membrane permeability of the molecules. At the end of the assay, the presence of free calcein or TDA in the supernatants is detected by direct fluorescence for calcein or by TR-FRET signal after addition of an europium solution resulting in the EuTDA chelate formation, respectively.
In this study, the nanoluciferase-expressing Raji and SKOV-3 cells were labeled with calcein or TDA, and ADCC was measured in simultaneous experiments using in parallel nanoluciferase-, calcein- or TDA-release read-outs. As described previously, a reference sample (100%) as well as under- (80%) and over- (125%) concentrated test samples were tested in each assay. With Raji cells, low to very low S/B ratios were obtained with calcein and TR-FRET methods compared with nanoluciferase (1.43 ± 0.27 and 1.15 ± 0.13 vs. 7.82 ± 1.03, respectively). These low ratios resulted in poorly reproducible results along the 3 independent experiments, as illustrated by the strong variations of the Emin, Emax and EC50 compared with the nanoluciferase parameters, and consistently with low correlation factors r2 ( and ). Average intra-replicate variations were good for TR-FRET (3.9%) and nanoluciferase (7.8%) methods, and a bit higher for calcein (16.4%). Furthermore, the apparent maximum lysis was completely aberrant, reaching more than 300% of specific lysis in some experiments. Despite the strong inter-assay asymptote variations with the calcein and TR-FRET methods, experimental biases were also calculated from the test samples and they consistently showed a poor to very poor accuracy of these 2 methods.
Table 3. Comparison of the characteristic parameters of ADCC assays using the nanoluciferase, calcein or TR-FRET methods in the unconstrained 4-parameter logistic regression model. The EC50 (means and the corresponding CV), Emin and Emax (mean and range) from the reference sample are indicated in the table for both cellular models, using the different readouts. Correlation coefficients (r2) are also indicated to illustrate the data fitting to the regression model. The relative biases (and ranges) obtained with the under- and over-concentrated test samples (80% and 125%) are also shown. The data were obtained from 3 independent experiments (see also ). * In the Raji/calcein model @125%, only one experiment over 3 gave data fitting with the logistic model.
Figure 6. Direct comparison of the nanoluciferase method to the calcein and TR-FRET methods. Nanoluciferase –expressing target cells (Raji or SKOV-3) were used to measure ADCC activities either directly by nanoluciferase-release read-out or after calcein or BATDA labeling. (A) The resulting percentages of specific lysis and unconstrained 4-parameter logistic regressions are shown for the reference Ab (100%, black circle and plain line) and for the under-concentrated (80%, open triangle and short-dotted line) and over-concentrated (125%, open square and long-dotted line) test samples. The upper and lower panels show the results obtained with the Raji cells (note the wide Y-axis range variations between the different methods) and with the SKOV-3 model, respectively, using the 3 methods (nanoluciferase on the left, calcein on the center, and TR-FRET on the right). Data are the means ± SD from 3 independent experiments. (B) After supernatant harvesting for nanoluciferase, calcein and TR-FRET signal measurements, SKOV-3 cell pellets were washed and stained for measuring the percentage of remaining living target cells by flow cytometry (see the Material and methods section for the analysis details). The resulting percentages are shown on each dot plot. Only the spontaneous release and the lowest and highest antibody concentrations are depicted on the figure. Data are from one experiment representative of 2. (C) The Emax parameters (from unconstrained 4-parameter logistic regressions) obtained in each assay (nanoluciferase, calcein or TR-FRET) using both read-out, i.e., the respective supernatant (SN) read-out (light gray bars) and the flow cytometry (FC) measurement on the pellets (black bars), are shown. Data are mean ± SD from 2 (flow cytometry) or 3 (supernatant reading) experiments. Using the nanoluciferase supernatant condition as a reference, Student's t tests were performed against the other conditions and results are shown on the graph: NS, not significant (p > 0.05); * p ≤ 0.05; ** p ≤ 0.005.
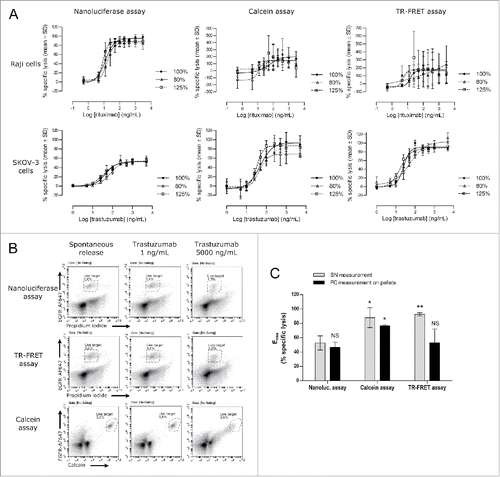
With the SKOV-3 cells, notwithstanding the low S/B ratios obtained with the calcein- and TR-FRET methods (1.63 ± 0.16% and 1.93 ± 0.05%, respectively, compared with 21.80 ± 2.59% with the nanoluciferase read-out), the results appeared more reproducible than in the Raji model, with global performances relatively close to the nanoluciferase method (see and ). Average intra-replicate variations were 4.5%, 2.4% and 8.2% for calcein, TR-FRET and nanoluciferase methods, respectively. Despite a comparable inter-assay precision between the 3 methods (illustrated by the EC50 coefficients of variation in the ), the nanoluciferase assay tends to exhibit a better accuracy (i.e., lower biases, in average) (). This last point was consistent with the better asymptote and slope similarities in the nanoluciferase assays between the reference and the test samples (and among the 3 experiments), compared with the calcein and TR-FRET methods. Indeed, for the nanoluciferase, calcein and TR-FRET methods, the maximum Emax CV were 6.4%, 23.5% and 12.9%, respectively, and maximum slope CV were 18.2%, 43.4% and 44.9%, respectively. More surprisingly, a major difference appeared between the methods in this model, since the SKOV-3 maximum lysis measured using the calcein or TR-FRET methods was found to be significantly higher (90–93% in average) than the maximum lysis measured using the nanoluciferase and 51Cr methods (35–53% in average, , , and ).
To determine the actual SKOV-3 target cell death percentage in these ADCC assay conditions, a flow cytometry-based approach was used, following a similar rationale than the data presented in the . At the end of the ADCC incubation period and after the supernatant harvesting for nanoluciferase, calcein or TR-FRET signal measurement, the cell pellets from the trastuzumab-based assay were resuspended, labeled with fluorescent anti-epidermal growth factor receptor (EGFR) antibody and analyzed by flow cytometry to measure the frequency of the residual living target cells (). The results, summarized in , showed that the specific lysis measured using the supernatant in the nanoluciferase assay (52.5 ± 2.9%, mean ± SD, n = 3) was consistent with the percentage of lysis measured by flow cytometry in both the nanoluciferase assay (46.6 ± 7.2%, mean ± SD, n = 2) and the TR-FRET assay (52.9 ± 19.2%, mean ± SD, n = 2) (p>0.05, Student t test). Hence, the maximum specific lysis measured in the supernatant of the TR-FRET assay appeared to be over-estimated (p = 0.0026 vs. nanoluciferase cytometry and p = 0.0372 vs. TR-FRET cytometry). Interestingly, in the calcein assay, both the supernatant signal measurement and the flow cytometry read-out showed a significantly increased specific lysis compared with the nanoluciferase assay supernatant measurement (93.2 ± 13.4%, p = 0.0200 and 76.7 ± 1.2%, p = 0.0222, respectively, mean ± SD and Student t test p value), while no significant difference could be found between these 2 read-outs of the calcein assay. This last result suggested an increased mortality specifically related to the calcein assay, competing with the ADCC-induced lysis. Overall, the nanoluciferase assay appeared as the most representative method for target cell cytotoxicity measurement, giving results consistent with flow cytometry and 51Cr-release methods.
Discussion
The availability of a biologically relevant and standardized bioassay technology for evaluating the cytotoxicity of therapeutic Abs and, more specifically, the ADCC activity is a key challenge in fulfilling regulatory authority requirements. To comply with the biological relevance objective, the technology should be based on an actual measurement of target cell death. To date, the most sensitive methods to evaluate target cell death are based on 51Cr (or other radionuclides, such as 111In) release measurement. However, the use and development of potency assays based on these methods, particularly in an industrial context, are strongly hampered by the constraints of radioactive element use.
Thus, we developed a non-radioactive method to measure cytotoxicity based on the transformation of target cells to stably express a reporter protein, whose release in the culture supernatant is correlated with the target cell death. Although this approach, which involves cell transformation and clone selection steps, may initially appear to be a fastidious and long process, it makes sense in the context of therapeutic Ab development and quality control. The few weeks required to establish a stable target cell clone are largely compensated by the substantially improved stability and reproducibility of the assay over the entire lifetime of the product, which may be 15 to 20 years, provided that careful characterization is performed, coupled with Master and Working cell bank-based management to optimize the assay stability over time. Moreover, with a judicious choice of the target cell line model, one transformed clone may be used for several candidate targets or antibodies, which increases the profitability of the initial investment.
In contrast to the previously published, and partially unsuccessful, approaches that involve firefly luciferase or β-galactosidase-expressing target cells,Citation39 we selected the recently marketed nanoluciferaseCitation36 as the reporter protein. Nanoluciferase has been used as a reporter gene for protein-ligand interactions and signaling pathway studiesCitation40,41 or has been coupled to a protein of interest for live imagingCitation42,43 and traffickingCitation44,45 studies. With a substantially increased half-life in extracellular medium compared with the firefly luciferase (7.7 d vs. 30 minutes, respectively),Citation36,39 nanoluciferase was compatible with the typical cytotoxicity assay durations. Furthermore, as a result of its high luminescence activity, the nanoluciferase-based assay may be performed using a very low number of target cells (500 cells/well). In contrast to β-galactosidase, nanoluciferase also appeared to be quickly and completely released during cell death, as illustrated by the perfect correlation with the flow cytometry cell death measurement in the apoptosis assays () and the fully comparable percentages of specific lysis with the 51Cr-release assay (, and ). Moreover, Schäffer et al.Citation39 identified significant differences between the percentages of specific lysis when the readout was 51Cr or β-galactosidase, and the latter always resulted in lower percentages. The mechanisms of ADCC and pore formation on the target cell membrane are complex and largely unresolved; however, our results suggest that the size of the reporter molecule matters. Lopez and colleaguesCitation46 recently demonstrated that small pores (120 to 170 Å) were transiently formed (during 20 to 80 seconds) in the immunological synapse to enable granzyme entry into the target cells. Since the preservation of membrane integrity during granzyme-induced apoptosis remains questionable,Citation47 these pores may also constitute an exit gate for the intracellular content during the ADCC process. Regardless of the involved release process, nanoluciferase, with its relatively small size (19 kDa), appears to be more easily released during the cytotoxicity process compared with bigger proteins, such as β-galactosidase (464 kDa) or the initially tested 47 kDa chimerical TR-FRET reporter protein.
This finding was further supported by our investigation of the release kinetics, which demonstrated a fast release of nanoluciferase, as quick as 2 h in the CDC model and 3 h for ADCC. This quicker release in the CDC assay was consistent with its mechanism of action, in which the lytic system was constituted by soluble factors available in excess, thereby leading to concomitant lysis, in contrast to the ADCC assay, in which the effector cells had to sequentially initiate cell-to-cell contacts before killing on a “first-come, first-served” basis. Consistently, the S/B ratios observed with the nanoluciferase method (between 7 and 15, depending on the assays and models) were comparable to classic S/B ratios observed with the 51Cr-release method (between 5 and 12 in similar conditions). Furthermore, the release kinetics and characteristics are comparable between both methods. When the ADCC and CDC assays were run in parallel in a first series of experiments comparing the nanoluciferase and 51Cr-release methods, using the same nanoluciferase-expressing target cells in the same experimental settings, the similarity between the 2 methods was confirmed, which resulted in highly similar characteristic curves and modeling parameters. Using the unconstrained model (), the EC50 CVs of the standard curves were relatively classical for these types of bioassays. Indeed, the majority of the assays exhibited between 15 and 25% CV variation, with the exception of 29% and 38% for the rituximab and trastuzumab 51Cr-based ADCC assays, respectively. The nanoluciferase assay performed generally better than 51Cr, with CVs that ranged from 7.2% to 22.8% and 18.6% to 38.4%, respectively.
Notwithstanding the impressive similarity between the 51Cr and nanoluciferase lysis measurements illustrated in and , a slight difference was identified in the rituximab (ADCC and CDC) and trastuzumab (ADCC) models regarding the EC50value, which was approximately 2-fold lower using the nanoluciferase readout compared with 51Cr. This result could not be explained by a difference in the ADCC activity of the sample because the same Ab preparation was used in each assay for both the 51Cr-labeled and non-labeled target cells. Thus, these findings suggested that despite comparable release characteristics, small differences may remain between the nanoluciferase and 51Cr release mechanisms in these models. Nevertheless, this small difference in the absolute EC50 values was not considered critical because the 51Cr- and nanoluciferase-determined EC50 were not supposed to be directly compared with each other, especially in a potency measurement-based approach.
Following the proof of concept that the nanoluciferase-release method was a valuable tool to efficiently measure cell death in a variety of experimental designs (e.g., apoptosis, ADCC and CDC assays), we attempted to assess its validation potential in a regulatory environment. According to the USP chapters <1032> and <1034>, the potency of a test sample may be measured relative to a reference sample by calculating their EC50 ratios in a constrained 4-parameter logistic model, provided that both curves in the unconstrained model are similar, i.e., have equivalent asymptotes and slopes (parallelism assumption). For each parameter, during assay development, the USP recommends determining equivalence margins representative of the assay variability by compiling historical data. This may be achieved, for example, by running the reference vs. itself, or vs. modified or altered batches whose potency has been demonstrated to be acceptable, for approximately 25 iterations.Citation48,49 However, the objective of our study was not to develop a specific potency assay for a given Ab product; rather, it was to provide preliminary experiments that illustrate the potentialities of the nanoluciferase cytotoxicity based method. Thus, we did not have access to a sufficient amount of historical data to accurately mark out the acceptable variability limits of asymptotes and slopes. Because of our limited set of data, we subsequently chose to diverge from the strict “relative potency” calculation described in the USP chapters and to use the EC50 ratio between the reference and standard samples from the unconstrained model as a surrogate evaluation of the relative potency. The unconstrained model was selected instead of the constrained model because, in general, constraining the model decreases the curve fitting to the experimental data points, which thus decreases the EC50 data representativeness. Using under- (80%) and over- (125%) concentrated preparations of the reference sample as test samples, we applied this analysis strategy to determine the accuracy and precision of the assay. To further provide robustness elements in this evaluation of performances, 2 different operators were involved in the 3 performed assays, and the assays were performed independently on separate days.
The EC50 ratios calculated for the test samples exhibited CVs that ranged from 9% to 36% () depending on the different conditions and models, consistent with the EC50 CVs calculated for the standard curves alone (), which ranged from 15% to 25%. Moreover, in general, the average experimental biases were inferior to 14%, with 2 exceptions at 23% (125% test sample in the SKOV-3 ADCC and 80% test sample in the Raji CDC). These values are classically observed or reported for these types of complex bioassays; however, they remain relatively high and may be improved in the future. This may be achieved by standardizing and optimizing the assay one step further, after identifying the criteria that critically affect the assay variability. For example, the preparation of target cells may be optimized by an assessment of the effect of several factors, such as the cell confluence before plating, the plating concentration and the time before effector cells or complement addition (e.g., to enable adherent cells to re-adhere). Even if a standardized effector cell population specifically developed to improve the ADCC assay reproducibility has been used in these experiments, optimization may likely be made on this part, for example, by adapting the effector:target (E:T) ratio to a specific target cell model.
We are conscious that the EC50 ratio-based analysis selected here does not follow the USP equivalence testing approach. In a “real life” assay development, equivalence margins will be defined on the basis of a substantial amount of historical data, which may enable the determination of accurate acceptance criteria, which are representative of the true assay variability. However, importantly, the nanoluciferase-based assay did not underperform compared with the 51Cr method and exhibited at least comparable, and sometimes better, performances compared with the historical method. In some models or conditions, 51Cr had a small advantage in terms of the CV or range; however, the nanoluciferase method performed better than 51Cr in other conditions. Thus, the validation potential of the nanoluciferase method appeared to be similar to the historical 51Cr method; moreover, it had substantially increased potentials regarding developability, particularly as a result of the absence of radioactivity, and long-term reproducibility, which resulted from the stably transformed target cell approach.
The last part of our study aimed to compare the new nanoluciferase method to some alternative non-radioactive methods for cytotoxicity measurement, i.e., the calcein-release method and the TR-FRET read-out (using the DELFIA® cytotoxicity kit from Perkin Elmer). Both the nanoluciferase-expressing Raji and SKOV-3 cell lines were tested for ADCC measurement after calcein or TDA-labeling, in parallel to the nanoluciferase release assay. The data suggested that neither the calcein nor the TDA labeling was adapted to the Raji cell model, the extremely low S/B ratio resulting in inconsistent and highly variable results. This might be related to a very high spontaneous release of calcein or TDA by the Raji cells, which is a common drawback of these 2 methods.Citation20,21 In contrast, the SKOV-3 cell line showed relatively good accuracy and precision performances that were very similar to the nanoluciferase method. However, despite these good performances, a major difference regarding the maximum lysis evaluation by the calcein and TR-FRET methods was observed. In all our previous experiments using 51Cr or nanoluciferase read-out, trastuzumab induced a partial lysis of the SKOV-3 target cells, rarely exceeding 50 to 60%, also consistent with some previously published data.Citation50 In contrast, maximum lysis appeared close to 100% with both the calcein and the TR-FRET methods.
We thus explored the residual percentage of living SKOV-3 cells at the end of the ADCC assay, using a flow cytometry approach targeting EGFR, another specific receptor of the SKOV-3 cells in this model. For the nanoluciferase and TR-FRET conditions, the living SKOV-3 cell gate targeted the PI-negative cells expressing EGFR at a bright to dim level, to take into account the previously reported EGFR downregulation in the presence of anti-HER2 antibodies.Citation51,52 For the calcein labeled cells, PI was not used because of its spectral overlapping with calcein, and the residual living SKOV-3 cells were gated on both the EGFR detection and the absence of calcein leakage. The data first demonstrated that the maximum lysis measured by the TR-FRET method on the culture supernatant was over-estimated, since the target cell specific lysis measured by flow cytometry was consistent with the nanoluciferase assay data. On the contrary, the flow cytometry data from the calcein-labeled cells were not significantly different from the supernatant's calcein signal measurement, reaching about 80 to 90% of specific lysis. Hence, consistently with previously published data,Citation53 our results suggest that the calcein labeling might be partially toxic for the target cells, resulting in an increased sensitivity to the Ab-induced cytotoxicity. Although these observations need to be widened in other cellular models, calcein and TR-FRET methods thus both appear inappropriate for ADCC testing in the tested models, while 51Cr- and nanoluciferase-release assays combine versatility, biological relevance and good assay performances.
Overall, the current findings demonstrate that the new nanoluciferase method is a valuable non-radioactive method that may replace the historical 51Cr-release assay for cytotoxicity measurement. It is safe, versatile, fast, easy to use and only requires simple equipment to perform. Its high sensitivity enables the use of only a very low number of target cells compared with other methods. Consequently, a limited number of effector cells is required per assay, which is particularly important, for example, in the case of patient-derived effector cells. These different advantages make the nanoluciferase method amenable to high throughput screening for limited operating costs. From a biological point of view, and in contrast to other existing non-radioactive methods, the nanoluciferase release is specific and fully correlated with target cell death. Moreover, the involved mechanisms appear to be comparable to the mechanisms involved in 51Cr release because good similarities were demonstrated between both methods in terms of the kinetics, spontaneous and maximum releases and specific lysis percentages in the ADCC and CDC models using adherent or non-adherent target cells. The full biological relevance of the nanoluciferase-mediated cell death measurement supports the regulatory recommendations and guidelines. Thus, the nanoluciferase method represents a suitable tool for the development of many cytotoxicity-related potency assays (e.g., ADCC, CDC, and apoptosis) required during the development of innovator or biosimilar therapeutic Abs, from early candidate screening to quality control testing for lot release.
Materials and methods
Cell lines and culture
All cultures were performed at 37°C in a humidified atmosphere that contained 5% CO2. The Raji (ECACC, catalog #85011429), SKOV-3 (ATCC, #HTB-77) and CHO/TNF cell lines were cultured in RPMI-1640 (Sigma-Aldrich, #R8758), McCoy's (Sigma-Aldrich, #M8403) and Ham's F12 (Sigma-Aldrich, #N6658) media, respectively, and were all supplemented with 10% fetal bovine serum (Sigma-Aldrich, #F7524). The CHO/TNF cells corresponded to a CHO-K1 cell line (ECACC, #85051005) that stably expressed a low-cleavage mutant form of TNF, deleted from amino acids 1 to 12,Citation54 which was previously developed in our laboratory (unpublished). Forty-eight hours before assay performance, target cells were passaged and cultured in their specified medium.
The cytotoxic T cells genetically modified to express a CD16/FcϵRIγ chimerical receptor were used as effector cells.Citation38 These cells enabled a significant improvement in the assay precision (unpublished). Eighteen to 24 h before the ADCC assay performance, the effector cells were thawed in RPMI-1640 that contained 50% fetal bovine serum (FBS; Sigma-Aldrich, F7524), centrifuged 10 minutes at 160 g and cultured in RPMI-1640 supplemented with 10% FBS and 300 IU/mL of IL-2 (Miltenyi Biotec, 130–093–903).
Construction of nanoluciferase-expressing cell lines
Vector construction
Using the nanoluciferase sequence available in GenBank (Accession Number JQ513379), chemical synthesis was performed after codon optimization for expression in eukaryotic cells using Invitrogen's GeneArt® and GeneOptimizer™ technologies.Citation55 The synthesized nanoluciferase sequence was subsequently sub-cloned under the control of the human CMV Immediate Early promoter in a PiggyBac vector that contained the puromycin resistance gene (SBI, PB510B-1) to obtain the pPB-NLuc vector.
Establishment and follow-up of the nanoluciferase cell lines
The Raji, SKOV-3 and CHO/TNF cell lines were electroporated with both pPB-NLuc and PiggyBac transposase (SBI, PB210PA-1) vectors using a Neon transfection system (Invitrogen, Carlsbad, CA) and were cultured in the presence of puromycin (Invivogen, #ant-pr) at 0.25, 5 and 20 µg/mL, respectively. After 7 to 10 d of antibiotic selection, transfectants were cloned by limiting dilution, and the clones were screened based on their nanoluciferase expression following the lysis of 3,000 cells with 0.24% Triton X-100 (Sigma-Aldrich, T9284) and the resulting luminescence was read as described below.
The selected clones were maintained in culture under antibiotic selection during 50 passages, and the nanoluciferase expression was regularly monitored by measuring the nanoluciferase S/B ratio after cell lysis. Briefly, 3,000 cells were incubated for 15 minutes in a 96-well flat-bottom microplate in culture medium alone or with 0.24% Triton X-100 to determine the spontaneous and maximum release, respectively. The incubation plates were subsequently centrifuged for 1 minute at 860 g, and 25 µL of supernatant were transferred in 96 half-area white microplates (Greiner BioOne, #675074). Twenty-five µL of substrate (NanoGlo® Luciferase assay system, Promega, #N1130) diluted 1/50 in phosphate-buffered saline (PBS; Sigma-Aldrich, D8537) were added to each well; after 3 minutes of incubation at room temperature, the plates were read with a Mithras LB940 multimode microplate reader (Berthold Technologies, Bad Wildbad, Germany) with an acquisition time of 0.05 s per well. The nanoluciferase activity was expressed in RLU. The S/B ratio was further calculated as the mean of the maximum release divided by the mean of the spontaneous release. The nanoluciferase-expressing clones were the only target cells used in this study.
Apoptosis induction
The apoptosis of nanoluciferase-expressing Raji cells (50,000 cells/well) was induced using incubation with increasing doses of staurosporine (Sigma-Aldrich, #S6942) (6.4 nM to 20 µM) or UV exposure (Benchtop UV Transilluminator STX-20-M, Uvitec, Cambridge, UK) for increasing periods (10 to 60 s) in a 100 µL final volume of RPMI-1640 10% FBS. Forty-eight hours after induction, 100 µL of RPMI-1640 in the presence or absence of 0.5% Triton X-100 were added to the wells. After 15 minutes at room temperature, the plates were centrifuged, and 25 µL of supernatant per well were used for the nanoluciferase signal measurement (as described previously). The percentage of dead cells was calculated for each condition as follows: (mean RLU of the cells in medium without Triton / mean RLU of the cells in medium with Triton) x 100.
The cell pellet was resuspended in the residual volume of medium and acquired on a BD Accuri™ C6 flow cytometer (BD Biosciences, San Jose, USA) following the addition of 0.25 µM TO-PRO-3 (Molecular Probes, #T3605). Living cells were gated based on the morphologic parameters (FSC/SSC) and TO-PRO-3 negativity, using the BD Accuri C6 software.
Antibodies
All therapeutic Abs were from commercially available stocks. Anti-CD20 rituximab (MabThera®, lot number H0146) and anti-HER2 trastuzumab (Herceptin®, lot number H4406) were purchased from Roche. Anti-TNF adalimumab (Humira®, lot number 34434) was purchased from Abbott Laboratories. For the ADCC and CDC assays, antibodies were serially diluted in RPMI-1640 to obtain the indicated concentrations. For the accuracy evaluation experiment, in addition to the standard reference range (considered the “reference sample”), additional ranges that represented 80% or 125% of the standard range nominal concentrations were prepared to mimic under- and over-potent samples (considered “test samples”) for accuracy evaluation.
51Cr and nanoluciferase comparison in ADCC and CDC assays
For the 51Cr-release assay, the nanoluciferase-expressing target cells were initially incubated in the presence of 1 µCi/106 cells of 51Cr (sodium chromate, PerkinElmer, NEZ030) for 1 h at 37°C and were then extensively washed before use to eliminate the excess free 51Cr. For the nanoluciferase-based assay, the nanoluciferase-expressing target cells were directly usable for the assay.
Except when otherwise stated, 3,000 target cells per well were plated in flat-bottom 96-well plates for all the ADCC and CDC assays, in a final volume of 100 µL, with each condition tested in triplicate. For the ADCC assay, the E:T ratio was 10:1, and the assay was performed in RPMI-1640 with 5% FBS. The CDC assays were performed in raw RMPI-1640 in the presence of a fixed quantity of complement per well, i.e., 1 CH50 (complement hemolytic activity 50)/well for Raji cells and 6 CH50/well for CHO/TNF cells, using serum from guinea pig titrated for complement activity (Sigma-Aldrich, #S1639).
For all assays, the target cells and antibodies were pre-incubated for 20 minutes at room temperature in the assay plate. Following the addition of effector cells or complement, the plates were incubated at 37°C, 5% CO2 for the indicated time (4 h, except when otherwise stated). Each measurement plate included maximum and spontaneous release conditions, i.e., the target cells in medium containing an irrelevant Ab (rituximab for SKOV-3 cells or trastuzumab for Raji and CHO/TNF cells), effector cells or complement, and the presence or absence of 0.01% Triton X-100, respectively. At the end of the incubation period, 50 µL of supernatants were transferred in 96 V-bottom microplates and centrifuged for 1 minute at 860 x g.
For the 51Cr-labeled cells, 25 µL of supernatant were subsequently transferred in Lumaplates (PerkinElmer). The plates were dried, protected from light, for one night at room temperature before gamma counting in a MicroBeta JET plate reader (PerkinElmer, Waltham, USA). For the unlabeled target cells, the nanoluciferase signal was read as described previously.
Nanoluciferase, calcein and TR-FRET methods comparison in ADCC assays
For the calcein- and TR-FRET-based assays, nanoluciferase-expressing Raji or SKOV-3 cells were labeled with calcein-AM (Biolegend, 425201) or with the fluorescence-enhancing ligand bis-(AM)-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (BATDA, DELFIA® EuTDA cytotoxicity kit, Perkin Elmer, AD0116). Briefly, 3×106 target cells were washed twice in RPMI-1640 and incubated 20 min at 37°C and at 106 cells/mL with calcein-AM (2.5 µM, according to preliminary labeling optimization experiments) or with BATDA (1/400 final dilution, according to the manufacturer's instructions). Cells were then washed 3 times with RPMI 10%FBS. For the nanoluciferase-based assay, the nanoluciferase-expressing target cells were directly usable for the assay.
The 3 different target cell populations (unlabeled, calcein-labeled and BATDA-labeled) were then run in parallel in an ADCC assay as described above in the “51Cr and nanoluciferase comparison in ADCC and CDC assays” section. At the end of the incubation period (4 h), 50 µL of each supernatant were transferred in 96 V-bottom microplates and centrifuged for 1 minute at 860 x g.
For the nanoluciferase-based assay, supernatants from the unlabeled target cells were then directly measured for nanoluciferase activity as described above. For the calcein-based assay, 25 µL of supernatants from calcein-labeled cells were transferred in a black 96-well half-area plate (Greiner Bio-One, 675076) and read for fluorescence (excitation 485 nm, emission 520 nm, reading time 0.3 s) on the Mithras LB940 reader. For the TR-FRET-based method, 20 µL of supernatants from BATDA-labeled cells were transferred in the provided 96-well flat-bottom plate and 200 µL of europium solution was added in each well and incubated 15 minutes under agitation before measurement, following the manufacturer's instructions. The TR-FRET signal was read on the LB940 reader with the following parameters: excitation 320 nm, emission 615 nm, counting time 1 s (500 cycles of 2 ms including a 50 µs delay and a 400 µs reading time).
In some experiments, the SKOV-3 cell pellets were harvested to measure the remaining living target cells by flow cytometry. Triplicate wells were first treated for 4 min with 50 µL of trypsin-EDTA (Sigma-Aldrich, T3924) to detach SKOV-3 target cells. Trypsin was then inactivated by the addition of 100 µL of RPMI-1640 10% FBS and plates were centrifuged 1 min at 500 g. After supernatants removal, the cell pellets were harvested and pooled in 100 µL of PBS 2% FBS. Cells were then labeled with AlexaFluor-647 coupled anti-EGFR antibody (BD Biosciences, 563577). After 20 min of incubation at 4°C, cells were washed twice with PBS 2% FBS and directly acquired on a C6 flow cytometer. For nanoluciferase and TR-FRET conditions, cells were acquired in the presence of 0.4 µg/mL of propidium iodide (PI, eBiosciences, 00–6990–50). The percentage of living SKOV-3 cells was determined in the PI-negative/EGFR positive gate, respectively, using the BD Accuri C6 software. For calcein conditions, because of the spectral leakage of the calcein fluorescence on the PI channels, the accurate measurement of PI-positive dead target cells was not possible and the frequency of residual target cells was only measured in the calcein-positive / EGFR-positive gate.
Data analysis
The data analysis was performed similarly for the 4 read-out methods. The percentage of specific lysis for each Ab concentration was calculated according to the following formula: [(experimental signal – mean spontaneous release) / (mean maximum release – mean spontaneous release)] x 100. For flow cytometry data, the percentage of dead cells was calculated as follows: 100 – [% of living cells].
The spontaneous and maximum release values were plate-specific. The data were modeled using an unconstrained 4-parameter logistic regression, which generated 4 independent output parameters for each Ab dilution range: EC50, maximal lysis (Emax), minimal lysis (Emin) and slope of the linear part. Furthermore, the percentage of spontaneous release was calculated as the ratio between the spontaneous and maximum signals x 100.
CVs, which is also referred to as the “relative standard deviation,” were calculated as [standard deviation]/[arithmetic mean].
To evaluate the performances of the methods, the EC50 ratios were calculated for each 80% and 125% test sample as [EC50(reference)]/[EC50(test)] from the corresponding unconstrained 4-parameter logistic regression models. For the accuracy evaluation, the experimental bias (in %) was subsequently calculated as an extrapolation of the USP <1033> formula: 100 x [(Measured EC50 ratio/Target EC50 ratio)-1]. The target EC50 ratios were 0.8 and 1.25 for the 80% and 125% test samples, respectively.
All statistical analyses, linear and nonlinear regressions and graphs were performed using GraphPad Prism® software. The specific lysis, S/B ratios and common calculations were performed using Microsoft Excel.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We are grateful to Pr. Gilles Thibault for his critical reading and helpful participation in the writing of this manuscript.
Funding
This work was performed in the frame of the “Fonds Unique Interministeriel” funded project PremiumADCC. We thank BPIfrance and Region Pays de la Loire for funding.
References
- EMA/CHMP/BWP/157653/07. Production and quality control of monoclonal antibodies and related substances [ Internet]. 2009 [cited 2016 Apr 21]; Available from: http://www.ema.europa.eu/ema/pages/includes/document/open_document.jsp?webContentId=WC500003074
- FDA. Points to consider in the manufacture and testing of monoclonal antibody products for human use [ Internet]. 1997 [cited 2016 Apr 21]; Available from: http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/OtherRecommendationsforManufacturers/UCM062750.pdf
- FDA. Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product. Guidance for Industry [ Internet]. 2015 [cited 2016 Apr 21]; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf
- FDA. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Guidance for Industry [ Internet]. 2015 [cited 2016 Apr 21]; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
- EMA/CHMP/437/04 Rev. 1. Similar biological medicinal products [ Internet]. 2014 [cited 2016 Apr 21]; Available from: http://www.ema.europa.eu/ema/pages/includes/document/open_document.jsp?webContentId=WC500176768
- ICH-Q2(R1). Text on validation of analytical procedures [ Internet]. In: International Conference on Harmonization, Geneva. 1994 [cited 2014 Jun 10]. page 1-5. Available from: http://www.ich.org/products/guidelines/quality/quality-single/article/validation-of-analytical-procedures-text-and-methodology.html
- US Pharmacopeial Convention. USP NF official text with revision bulletins [ Internet]. [cited 2017 Jan 19]; Available from: http://www.usp.org/usp-nf/official-text
- Congy-Jolivet N, Probst A, Watier H, Thibault G. Recombinant therapeutic monoclonal antibodies: mechanisms of action in relation to structural and functional duality. Crit Rev Oncol Hematol 2007; 64:226-33; PMID:17716905; http://dx.doi.org/10.1016/j.critrevonc.2007.06.013
- Dall'Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, Watier H, Thibault G. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res 2004; 64:4664-9; PMID:15231679; http://dx.doi.org/10.1158/0008-5472.CAN-03-2862
- Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002; 99:754-8; PMID:11806974; http://dx.doi.org/10.1182/blood.V99.3.754
- Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol 2008; 26:1789-96; PMID:18347005; http://dx.doi.org/10.1200/JCO.2007.14.8957
- Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, Chang H-M, Borucka E, Lurje G, Sherrod AE, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor–expressing metastatic colorectal cancer patients treated with single-agent Cetuximab. J Clin Oncol 2007; 25:3712-8; PMID:17704420; http://dx.doi.org/10.1200/JCO.2006.08.8021
- Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, Theriault J, Crocker L, Pabonan O, Baginski T, Meng G, et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res 2010; 70:4481-9; PMID:20484044; http://dx.doi.org/10.1158/0008-5472.CAN-09-3704
- Mossner E, Brunker P, Moser S, Puntener U, Schmidt C, Herter S, Grau R, Gerdes C, Nopora A, van Puijenbroek E, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010; 115:4393-402; PMID:20194898; http://dx.doi.org/10.1182/blood-2009-06-225979
- Sehn LH, Chua NS, Mayer J, Dueck GS, Trněny M, Bouabdallah K, Fowler NH, Delwail V, Press OW, Salles GA, et al. GADOLIN: Primary results from a phase III study of obinutuzumab plus bendamustine compared with bendamustine alone in patients with rituximab-refractory indolent non-Hodgkin lymphoma. ASCO Meeting Abstracts 2015; 33:LBA8502.
- Cartron G, Guibert S de, Dilhuydy MS, Morschhauser F, Leblond V, Dupuis J, Mahe B, Bouabdallah R, Lei G, Wenger M, et al. Obinutuzumab (GA101) in relapsed/refractory chronic lymphocytic leukemia: final data from the phase 1/2 GAUGUIN study. Blood 2014; 124:2196-202; PMID:25143487; http://dx.doi.org/10.1182/blood-2014-07-586610
- Reichert JM. Antibodies to watch in 2014. mAbs 2014; 6:5-14; PMID:24284914; http://dx.doi.org/10.4161/mabs.27333
- Sanderson AR. Quantitative titration, kinetic behaviour, and inhibition of cytotoxic mouse isoantisera. Immunology 1965; 9:287-300; PMID:5838200
- Brunner KT, Mauel J, Cerottini JC, Chapuis B. Quantitative assay of the lytic action of immune lymphoid cells of 51Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology 1968; 14:181-96; PMID:4966657
- Lichtenfels R, Biddison WE, Schulz H, Vogt AB, Martin R. CARE-LASS (calcein-release-assay), an improved fluorescence-based test system to measure cytotoxic T lymphocyte activity. J Immunol Methods 1994; 172:227-39; PMID:7518485; http://dx.doi.org/10.1016/0022-1759(94)90110-4
- Granberg C, Blomberg K, Hemmilä I, Lövgren T. Determination of cytotoxic T lymphocyte activity by time-resolved fluorometry using europium-labelled concanavalin A-stimulated cells as targets. J Immunol Methods 1988; 114:191-5; PMID:2972782; http://dx.doi.org/10.1016/0022-1759(88)90173-1
- Somanchi SS, McCulley KJ, Somanchi A, Chan LL, Lee DA. A novel method for assessment of natural killer cell cytotoxicity using image cytometry. PLoS One 2015; 10:e0141074; PMID:26492577; http://dx.doi.org/10.1371/journal.pone.0141074
- Flieger D, Gruber R, Schlimok G, Reiter C, Pantel K, Riethmüller G. A novel non-radioactive cellular cytotoxicity test based on the differential assessment of living and killed target and effector cells. J Immunol Methods 1995; 180:1-13; PMID:7897241; http://dx.doi.org/10.1016/0022-1759(94)00293-6
- Wilkinson RW, Lee-MacAry AE, Davies D, Snary D, Ross EL. Antibody-dependent cell-mediated cytotoxicity: a flow cytometry-based assay using fluorophores. J Immunol Methods 2001; 258:183-91; PMID:11684135; http://dx.doi.org/10.1016/S0022-1759(01)00474-4
- Sheehy ME, McDermott AB, Furlan SN, Klenerman P, Nixon DF. A novel technique for the fluorometric assessment of T lymphocyte antigen specific lysis. J Immunol Methods 2001; 249:99-110; PMID:11226468; http://dx.doi.org/10.1016/S0022-1759(00)00329-X
- Korzeniewski C, Callewaert DM. An enzyme-release assay for natural cytotoxicity. J Immunol Methods 1983; 64:313-20; PMID:6199426; http://dx.doi.org/10.1016/0022-1759(83)90438-6
- Corey MJ, Kinders RJ, Brown LG, Vessella RL. A very sensitive coupled luminescent assay for cytotoxicity and complement-mediated lysis. J Immunol Methods 1997; 207:43-51; PMID:9328585; http://dx.doi.org/10.1016/S0022-1759(97)00098-7
- Cree IA, Andreotti PE. Measurement of cytotoxicity by ATP-based luminescence assay in primary cell cultures and cell lines. Toxicol Vitro 1997; 11:553-6; PMID:20654351; http://dx.doi.org/10.1016/S0887-2333(97)00060-X
- Peper JK, Schuster H, Löffler MW, Schmid-Horch B, Rammensee H-G, Stevanović S. An impedance-based cytotoxicity assay for real-time and label-free assessment of T-cell-mediated killing of adherent cells. J Immunol Methods 2014; 405:192-8; PMID:24486140; http://dx.doi.org/10.1016/j.jim.2014.01.012
- Seidel UJE, Vogt F, Grosse-Hovest L, Jung G, Handgretinger R, Lang P. γδ T cell-mediated antibody-dependent cellular cytotoxicity with CD19 antibodies assessed by an impedance-based label-free real-time cytotoxicity assay. Front Immunol 2014; 5:618; PMID:25520723; http://dx.doi.org/10.3389/fimmu.2014.00618
- Parekh BS, Berger E, Sibley S, Cahya S, Xiao L, LaCerte MA, Vaillancourt P, Wooden S, Gately D. Development and validation of an antibody-dependent cell-mediated cytotoxicity-reporter gene assay. MAbs 2012; 4:310-8; PMID:22531445; http://dx.doi.org/10.4161/mabs.19873
- Wolint P, Betts MR, Koup RA, Oxenius A. Immediate cytotoxicity but not degranulation distinguishes effector and memory subsets of CD8+ T Cells. J Exp Med 2004; 199:925-36; PMID:15051762; http://dx.doi.org/10.1084/jem.20031799
- de Romeuf C, Dutertre C, Le Garff-Tavernier M, Fournier N, Gaucher C, Glacet A, Jorieux S, Bihoreau N, Behrens C, Beliard R, et al. Chronic lymphocytic leukaemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcgammaRIIIA/CD16. Br J Haematol 2008; 140:635-43; PMID:18302712; http://dx.doi.org/10.1111/j.1365-2141.2007.06974.x
- Repp R, Kellner C, Muskulus A, Staudinger M, Nodehi SM, Glorius P, Akramiene D, Dechant M, Fey GH, van Berkel PHC, et al. Combined Fc-protein- and Fc-glyco-engineering of scFv-Fc fusion proteins synergistically enhances CD16a binding but does not further enhance NK-cell mediated ADCC. J Immunol Methods 2011; 373:67-78; PMID:21855548; http://dx.doi.org/10.1016/j.jim.2011.08.003
- Kawaguchi Y, Kono K, Mizukami Y, Mimura K, Fujii H. Mechanisms of escape from Trastuzumab-mediated ADCC in esophageal squamous cell carcinoma: relation to susceptibility to perforin-granzyme. Anticancer Res 2009; 29:2137-46; PMID:19528474
- Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol 2012; 7:1848-57; PMID:22894855; http://dx.doi.org/10.1021/cb3002478
- Mamidi S, Cinci M, Hasmann M, Fehring V, Kirschfink M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol Oncol 2013; 7:580-94; PMID:23474221; http://dx.doi.org/10.1016/j.molonc.2013.02.011
- Clémenceau B, Congy-Jolivet N, Gallot G, Vivien R, Gaschet J, Thibault G, Vié H. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood 2006; 107:4669-77; PMID:16514054; http://dx.doi.org/10.1182/blood-2005-09-3775
- Schäfer H, Schäfer A, Kiderlen AF, Masihi KN, Burger R. A highly sensitive cytotoxicity assay based on the release of reporter enzymes, from stably transfected cell lines. J Immunol Methods 1997; 204:89-98; PMID:9202713; http://dx.doi.org/10.1016/S0022-1759(97)00040-9
- Ho P, Yue K, Pandey P, Breault L, Harbinski F, McBride AJ, Webb B, Narahari J, Karassina N, Wood KV, et al. Reporter enzyme inhibitor study to aid assembly of orthogonal reporter gene assays. ACS Chem Biol 2013; 8:1009-17; PMID:23485150; http://dx.doi.org/10.1021/cb3007264
- Zhang L, Song G, Xu T, Wu QP, Shao XX, Liu YL, Xu ZG, Guo ZY. A novel ultrasensitive bioluminescent receptor-binding assay of INSL3 through chemical conjugation with nanoluciferase. Biochimie 2013; 95:2454-9; PMID:24056075; http://dx.doi.org/10.1016/j.biochi.2013.09.008
- Stacer AC, Nyati S, Moudgil P, Iyengar R, Luker KE, Rehemtulla A, Luker GD. NanoLuc reporter for dual luciferase imaging in living animals. Mol Imaging 2013; 12:1-13; PMID:24371848
- Tran V, Moser LA, Poole DS, Mehle A. Highly sensitive real-time in vivo imaging of an influenza reporter virus reveals dynamics of replication and spread. J Virol 2013; 87:13321-9; PMID:24089552; http://dx.doi.org/10.1128/JVI.02381-13
- Song G, Jiang Q, Xu T, Liu YL, Xu ZG, Guo ZY. A convenient luminescence assay of ferroportin internalization to study its interaction with hepcidin. FEBS J 2013; 280:1773-81; PMID:23413836; http://dx.doi.org/10.1111/febs.12192
- Norisada J, Hirata Y, Amaya F, Kiuchi K, Oh-Hashi K. A sensitive assay for the biosynthesis and secretion of MANF using NanoLuc activity. Biochem Biophys Res Commun 2014; 449:483-9; PMID:24845376; http://dx.doi.org/10.1016/j.bbrc.2014.05.031
- Lopez JA, Susanto O, Jenkins MR, Lukoyanova N, Sutton VR, Law RHP, Johnston A, Bird CH, Bird PI, Whisstock JC, et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 2013; 121:2659-68; PMID:23377437; http://dx.doi.org/10.1182/blood-2012-07-446146
- Cullen SP, Martin SJ. Mechanisms of granule-dependent killing. Cell Death Differ 2007; 15:251-62; PMID:17975553; http://dx.doi.org/10.1038/sj.cdd.4402244
- Saama PM. A unified approach for performing an analytical methods comparability study [ Internet]. 2015 [cited 2016 Apr 21]; Available from: http://www.cbinet.com/conference/agenda/pi15081
- Hauck WW, Capen RC, Callahan JD, Muth JED, Hsu H, Lansky D, Sajjadi NC, Seaver SS, Singer RR, Weisman D. Assessing parallelism prior to determining relative potency. PDA J Pharm Sci Technol 2005; 59:127-37; PMID:15971545
- Naruse I, Fukumoto H, Saijo N, Nishio K. Enhanced anti-tumor effect of trastuzumab in combination with Cisplatin. Japanese J Cancer Res 2002; 93:574-81; PMID:12036454; http://dx.doi.org/10.1111/j.1349-7006.2002.tb01293.x
- Wehrman TS, Raab WJ, Casipit CL, Doyonnas R, Pomerantz JH, Blau HM. A system for quantifying dynamic protein interactions defines a role for Herceptin in modulating ErbB2 interactions. PNAS 2006; 103:19063-8; PMID:17148612; http://dx.doi.org/10.1073/pnas.0605218103
- Kuwada SK, Scaife CL, Kuang J, Li X, Wong RF, Florell SR, Coffey RJ, Gray PD. Effects of trastuzumab on epidermal growth factor receptor-dependent and -independent human colon cancer cells. Int J Cancer 2004; 109:291-301; PMID:14750183; http://dx.doi.org/10.1002/ijc.11686
- Liminga G, Nygren P, Dhar S, Nilsson K, Larsson R. Cytotoxic effect of calcein acetoxymethyl ester on human tumor cell lines: drug delivery by intracellular trapping. Anticancer Drugs 1995; 6:578-85; PMID:7579562; http://dx.doi.org/10.1097/00001813-199508000-00011
- Perez C, Albert I, DeFay K, Zachariades N, Gooding L, Kriegler M. A nonsecretable cell surface mutant of tumor necrosis factor (TNF) kills by cell-to-cell contact. Cell 1990; 63:251-8; PMID:2208285; http://dx.doi.org/10.1016/0092-8674(90)90158-B
- Fath S, Bauer AP, Liss M, Spriestersbach A, Maertens B, Hahn P, Ludwig C, Schäfer F, Graf M, Wagner R. Multiparameter RNA and codon optimization: A standardized tool to assess and enhance autologous mammalian gene expression. PLoS One 2011; 6:e17596; PMID:21408612; http://dx.doi.org/10.1371/journal.pone.0017596