Abstract
After nearly a decade without growth in atmospheric methane, there are indications of renewed growth from 2007. Reports of this renewal portray it as global in extent, and due wholly or largely to growth in emissions. Surface methane mixing ratios and constituent δ13C values have been measured approximately twice monthly at Baring Head, New Zealand (41°S, 175°E) since 1989. Surface mixing ratios have been measured continuously at Lauder, New Zealand (45°S, 170°E) since 2007. Also at Lauder, tropospheric-mean mole fractions of methane have been retrieved from ground-based near-infrared solar spectra since 2004. These mixing ratio datasets are consistent with growth rates of about 7.5 and 4.9 ppb year−1 during 2007 and 2008. We consider the possible origins of this growth based on their imprint on δ13C values.
Keywords:
Introduction
The atmospheric methane (CH4) burden approximately doubled during the twentieth century, largely due to emissions from human activities (Dlugokencky et al. Citation1994b; Cunnold et al. Citation2002). Following a strong growth of ∼16 ppb year−1 during 1978–1987 (Blake and Rowland Citation1988), the growth rate slowed thereafter (Steele et al. Citation1992; Dlugokencky et al. Citation1994a, Citation1998; Cunnold et al. Citation2002; Simpson et al. Citation2002) compatibly with a decade-long approach toward steady state (Dlugokencky et al. Citation2003). However, a renewal in growth approaching 10 ppb year−1 has been reported for 2007 followed by a lower growth for 2008 (Rigby et al. Citation2008; Dlugokencky et al. Citation2009).
These documented changes in atmospheric methane have yet to be fully and quantitatively explained in terms of changes to CH4 sources and sinks. Geographic information on CH4 mixing ratios has helped to identify the latitudinal origins of changes, usually in emissions. Such studies have attributed much of the inter-annual variability during the 1990s to variable wetland emissions (Bousquet et al. Citation2006), and have raised the possibility of reduced emissions from natural gas systems in territories of the former Soviet Union during the early 1990s (Dlugokencky et al. Citation1994a, Citation2003). In addition, isotopic measurements of the 13C/12C isotope ratio in methane (δ13C(CH4)) have been used to diagnose the global CH4 budget and changes to it (e.g. Stevens and Engelkemeir Citation1988; Lowe et al. Citation1994; Quay et al. Citation1999; Lassey et al. Citation2007; Tyler et al. Citation2007). Such diagnoses rely on distinctive associations of δ13C with CH4 origin, particularly distinguishing biogenic sources (typical δ13C ≈ −60‰) from pyrogenic sources (typical δ13C ≈ −25‰) and fossil sources (typical δ13C ≈ −40‰), while also accounting for isotope fractionation of sink processes. However, long-term isotope records are geographically sparse.
Methane mixing ratio (MR) and associated δ13C values have been measured in flask samples from the coastal site of Baring Head, New Zealand (BHD: 41°S, 175°E) since 1989; the record for δ13C(CH4) is the longest reported in the world that is both of high frequency (twice-monthly with higher frequency early in the record) and representative of broad-scale air masses. In addition, we report two CH4 MR records acquired with high temporal resolution at the Total Carbon Column Observing Network (TCCON) inland site of Lauder, New Zealand (LDR: 45°S, 170°E), 585 km to the south-west of BHD. The LDR records are based on retrievals from Fourier transform spectrometry (FTS), one at the surface using closed-cell FTS commenced in 2007, and the other from upward-looking near-infrared (NIR) solar FTS since 2004. We examine these datasets for recent changes to the growth rate as reported by Rigby et al. (Citation2008) and by Dlugokencky et al. (Citation2009).
The NIWA datasets
BHD samples are collected during periods of stable CO2 and wind speeds exceeding 7.7 m s−1, and their origin from south-westerly oceanic air masses is confirmed by 5-day back-trajectories. All MRs are determined by gas chromatographic analysis of dried samples against recognized standards calibrated on the NOAA04 scale (Dlugokencky et al. Citation2005). All δ13C values are measured following cryogenic isolation by dual-inlet mass spectrometry against standards calibrated against Vienna Peedee Belemnite (VPDB) through IAEA references NBS19 and LSVEC (Lowe et al. Citation1991, Citation1994). Approximately monthly samples collected at the Antarctic Specially Protected Area at Arrival Heights (ASPA122), near Scott Base, Antarctica (AHT: 78°S, 167°E) are analyzed by the same methods following storage in Antarctica until late summer. The AHT datasets (not shown here) differ little from BHD and support the findings reported below.
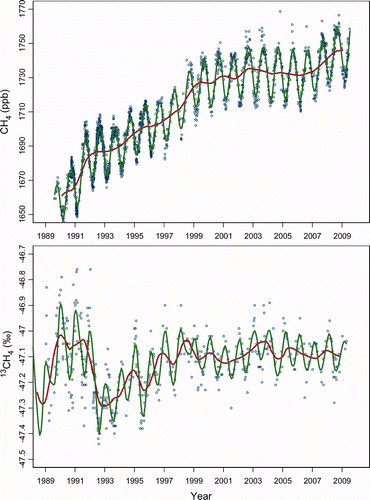
The BHD dataset has been subjected to seasonal-trend decomposition based on Loess filtering (STL) (Cleveland et al. Citation1990). This procedure segregates a smooth trend from a seasonal component that is not harmonically constrained and may change slowly with time, leaving residuals that can be examined for recurring features (versus random departures) or patterns. shows each data record together with the decomposed seasonality and trend. Note that this decomposition differs from that reported for NOAA/ESRL data which applies a quadratic trend plus a 4-harmonic fit (e.g. Steele et al. Citation1992; Dlugokencky et al. Citation1994a, Citation1994b, Citation1998). Thus, detailed comparisons between these differing decompositions can mislead.
Figure 1. Time series of MR, August 1989 to August 2009 (upper panel: NOAA04 scale), and δ13C, December 1987 to April 2009 (lower panel: VPDB scale) for methane measured at Baring Head, New Zealand. Data series (blue circles) are decomposed into trend (red) and seasonality (green) as described in the text. Tick marks on the abscissas denote 1 January of the particular year.
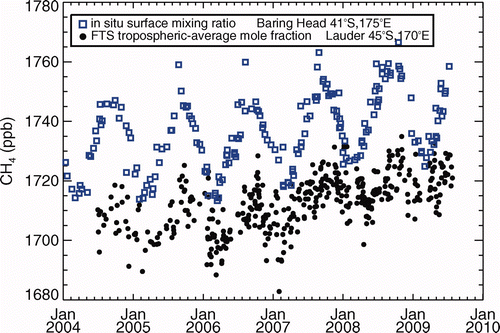
At LDR, the tropospheric-average dry air mole fraction (TAMF) of CH4 (essentially, the pressure-weighted average MR) is derived from NIR solar spectra using the algorithm of Washenfelder et al. (Citation2003) which estimates the stratospheric contribution to the CH4 total column (derived from HF) and a simultaneous retrieval of total column O2 to infer the CH4 TAMF. CH4 retrievals were performed using the revised spectroscopic line parameters of Frankenberg et al. (Citation2008). Absolute calibration of FTS measurements to the NOAA04 scale will be achieved through instrumented aircraft overpasses of the Lauder site during the HIAPER Pole-to-Pole Observation Experiment. The daily-average TAMFs for CH4 shown in are not yet calibrated to the NOAA04 scale.
Figure 2. Daily mean tropospheric-average CH4 dry air mole fraction (TAMF) retrieved from NIR solar absorption spectra acquired by Fourier Transform Infrared Spectrometry at Lauder, compared with gas chromatographic analysis of flask samples from Baring Head. The Lauder TAMF dataset has yet to be calibrated, which is expected to account for the mean offset between the datasets.
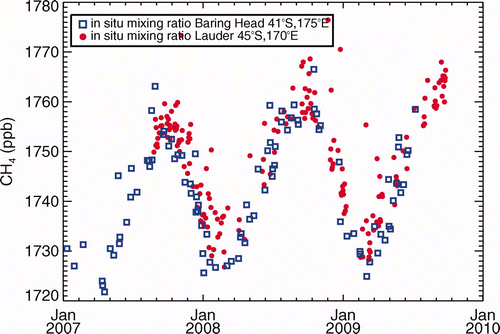
Surface concentrations of TCCON target gases at LDR employ a second, closed-path FTS in collaboration with the University of Wollongong, Australia. Trace gas number densities are retrieved from white cell transmission spectra (10-min integration) of dried air samples and combined with precise measurements of cell pressure and temperature to determine the gas MRs (Griffith Citation1996; Esler et al. Citation2000). A daily calibration cycle relates retrieved CH4 MRs to the NOAA04 scale. reports LDR measurements that are representative of regional scales (well mixed boundary layer), based on time of day (15:00 h–16:00 h local time) and surface windspeed (>6 m s−1). BHD data for the same period are included in for comparison.
Figure 3. Screened hourly mean surface CH4 MR (15:00 h–16:00 h local time, |u| > 6 m s−1) representative of regional scales derived from continuous closed-cell FTS measurements at Lauder, New Zealand, compared with flask samples from Baring Head analyzed by gas chromatography.
Discussion
Baring Head dataset
The CH4 MR record at BHD () is comparable to others reported for the southern hemisphere (SH) sites both in the NOAA/ESRL network (http://www.esrl.noaa.gov/gmd/ccgg) and, to early 2008, in the AGAGE and CSIRO networks that include Cape Grim, Australia, a SH site of similar latitude to BHD (Rigby et al. Citation2008). In the extra-tropical SH, CH4 seasonality is dominated by in situ removal by hydroxyl (OH) resulting in a very regular seasonal cycle, whereas northern hemisphere (NH) datasets display more complex seasonal structure due to the compounding effects of seasonal sources and transport (Dlugokencky et al. Citation1994b; Cunnold et al. Citation2002).
According to the STL decomposition, CH4 MR at BHD exhibits the following trend pattern: a steady growth averaging 6.3 ppb year−1 over calendar years 1993–1998; a slowed growth rate averaging 2.6 ppb year−1 over 1999–2002; a slightly negative growth rate –0.3 ppb year−1 over 2003–2006; and renewed growth of 7.5 and 4.9 ppb year−1 over 2007 and 2008, respectively. Taken together, the de-seasonalized MR of 1746 ppb at BHD in January 2009 is 21 ppb higher than 10 years earlier, with more than half of that growth occurring during 2007–2008.
Rigby et al. (Citation2008) report a growth of “almost 10 ppb at all locations” in their network for 2007 while Dlugokencky et al. (Citation2009) assess growth at 8.3 ± 0.6 ppb globally and 9.2 ± 0.3 ppb for the SH based on their more extensive NOAA network. The 2007 growth of 7.5 ppb year−1 at BHD is consistent with these estimates, recognizing the distinct analytical approaches to seasonal-trend decomposition. The 2008 growth of 4.9 ppb at BHD also supports the global assessment of 4.4 ± 0.6 ppb (Dlugokencky et al., Citation2009) with SH growth 3.7 ppb (E.J. Dlugokencky, personal communication).
compares the growth rates at BHD and LDR centered on calendar years 2005–2008, without the “stiffness” requirement of the STL procedure.
The most striking feature of the BHD δ13C record is the abrupt change in 1992 (). Although near-contemporaneous with the Mt Pinatubo eruption, that feature remains unexplained despite detailed scrutiny (Lowe et al. Citation1997; Mak et al. Citation2000). Of more relevance here is the absence of a prominent feature that could be associated with the recent increase in growth rate. After 1993, annual-mean δ13C steadily rises at a slowing rate as if approaching a steady state near −47.1‰, with fluctuations that include a small rise by ∼0.1‰ over 2001–2004 followed by a fall of comparable magnitude since 2004 that may not extend beyond 2007. A superimposed seasonal cycle has varying amplitude that may be smaller in recent years. However, there is no unequivocal feature accompanying the marked 2007 renewal in atmospheric growth. This could suggest that the cause of the renewed growth possesses minimal isotopic leverage. However, noting the slow response of atmospheric δ13C to changes in sources (Tans 1997), it is likely too early for such a renewal to have imposed itself discernibly on the isotope record.
The seasonality in both MR and δ13C at BHD are largely controlled by the seasonality of CH4 removal, with amplitude in δ13C that is proportional to the strength of the isotopic fractionation in the seasonal component of all removal processes combined (the “seasonal sink”) (Allan et al. Citation2001). Put mathematically after Allan et al. (Citation2001), if ΔC/Co denotes the seasonal amplitude in the (de-trended) MR, relative to the annual mean, and Δδ is the seasonal amplitude in (de-trended) δ13C, then to a good approximation
An annualized time series for ϵ at BHD calculated by Allan et al. (Citation2005) for calendar years 1992–2003 varies between −6 and −17‰, which is attributed to an inter-annually varying role of the chlorine sink. In extending the analysis through to 2008, ϵ shows remarkably less variability than was evident in the 1990s: |ϵ| falls from an average of 8.8 ± 1.6‰ in 2001–2004 to an average of 5.7 ± 1.7‰ throughout 2005–2007, only to rise again to 9.8 ± 3.1‰ in 2008. A weakened chlorine sink would by itself initiate a downward trend in δ13C as well as a reduced seasonal amplitude in δ13C through a lower |ϵ|. Such a weakening during 2005–2007 could partially account for the downward trend and reduced amplitude observed for δ13C at BHD at that time, and a strengthening during 2008 accounts for a reversal of those features. Chlorine sink changes would impact minimally on MR (and, for 2008, in the direction opposite from observation), whereas small OH changes would affect MR more than ϵ. It therefore seems unlikely that the observed 2007–2008 CH4 growth is primarily sink controlled, as Dlugokencky et al. (Citation2009) also find. However, usage of local ϵ to interpret large-scale changes to CH4 budget is speculative until sink seasonality is better characterized.
The lack of features in the BHD δ13C record that are synchronous with CH4 growth is most easily explained by multiple budget changes, such as a distributed mix of source increases with a weighted mean δ13C near the source mean of ∼−54‰. This could implicate a balance between higher biogenic emissions and a limited increase in pyrogenic or fossil emissions. Alternatively, CH4 from tropical wetlands is among the most 13C-enriched of biogenic sources (Bréas et al. Citation2001, and references therein) with δ13C close to that source mean. Because tropical wetlands south of the equator are a major component of SH emissions, a strong emission increase from tropical wetlands during 2007 could shed light on the interesting finding by Dlugokencky et al. (Citation2009) that the mean SH CH4 growth in 2007 (9.2 ppb) exceeded NH growth (7.3 ppb) despite the highest zonal growth being in high northern latitudes (13.7 ppb north of 53°N). Nevertheless, because δ13C responds slowly to source changes (Tans 1997), the constraint of a source mix near −54‰ is quite weak.
Lauder dataset
Although of more recent commencement, the LDR records generally confirm the representativeness of BHD MRs over the NZ region. Atmospheric CH4 growth rates centered on calendar years 2005–2008 based on column retrievals at LDR since 2004 compare favorably with those based on analyses of surface air at BHD (), and are in good accord also with STL analysis of the latter record (Section 3.1). The high-frequency surface FTS measurements at LDR corroborate the BHD MR record from 2007 (), including the lower growth in 2008 than in 2007 that is inferred both at BHD and elsewhere (Dlugokencky et al. Citation2009). While there is a hint of possible systematic differences between the BHD and LDR surface records during some seasons, the latter record is as yet too short for detailed trend and seasonal analysis.
Table 1. Growth rate by calendar-year (ppb year−1).
Conclusions
Our NZ methane data generally confirm the 2007 growth spurt described by Rigby et al. (Citation2008) and by Dlugokencky et al. (Citation2009), followed by a lower growth rate in 2008. Additionally, our unique δ13C(CH4) record suggests that the causes of these changes probably carry little isotope leverage that is yet discernible, which would be consistent with: (i) increased biogenic emissions along with either pyrogenic or fossil emissions; (ii) emission growth from southern tropical wetlands; or (iii) a lag between an emission increase and the associated isotopic response. Sink changes are not indicated by our data as drivers of the renewed growth, but may nevertheless include a weakening of the chlorine sink, 2005–2007, whose role is often overlooked in quantitative assessments of the global CH4 budget.
Acknowledgments
Dave Lowe was instrumental in setting up the measurement program and quality-control systems at Baring Head. The TCCON team at Lauder is acknowledged for the quality-controlled datasets collected there. Geoff Toon and Jean-Francois Blavier (JPL) developed the FFT and retrieval software for the TCCON. Discussion with Ed Dlugokencky (NOAA/ESRL, Boulder, CO) has provided a global context for the data reported herein. This work is supported by the New Zealand Foundation for Research, Science and Technology under contracts C01X0703 and C01X0406. Antarctica New Zealand supported and assisted with the Arrival Heights air sampling program.
Related Research Data
References
- Allan , W , Manning , M R , Lassey , K R , Lowe , D C and Gomez , A J . 2001 . Modeling the variation of δ13C in atmospheric methane: phase ellipses and the kinetic isotope effect . Global Biogeochem Cycles , 15 ( 2 ) : 467 – 481 .
- Allan , W , Lowe , D C , Gomez , A J , Struthers , H and Brailsford , G W . 2005 . Interannual variation of 13C in tropospheric methane: implications for a possible atomic chlorine sink in the marine boundary layer . J Geophys Res , 110 : D11306 doi: 10.1029/2004JD005650
- Allan , W , Struthers , H and Lowe , D C . 2007 . Methane carbon isotope effects caused by atomic chlorine in the marine boundary layer: global model results compared with southern hemisphere measurements . J Geophys Res , 112 : D04306 doi: 10.1029/2006J D007369
- Blake , D R and Rowland , F S . 1988 . Continuing worldwide increase in tropospheric methane, 1978–1987 . Science , 239 : 1129 – 1131 .
- Bousquet , P , Ciais , P , Miller , J B , Dlugokencky , E J , Hauglustaine , D A , Prigent , C , van der Werf , G R , Peylin , P , Brunke , E-G Carouge , C . 2006 . Contribution of anthropogenic and natural sources to atmospheric methane variability . Nature , 443 : 439 – 443 .
- Bréas , O , Guillou , C , Reniero , F and Wada , E . 2001 . The global methane cycle: isotopes and mixing ratios, sources and sinks . Isotopes Environ Health Studies , 37 : 257 – 379 .
- Cantrell , C A , Shetter , R E , McDaniel , A H , Calvert , J G , Davidson , J A , Lowe , D C , Tyler , S C , Cicerone , R J and Greenberg , J P . 1990 . Carbon kinetic isotope effect in the oxidation of methane by the hydroxyl radical . J Geophys Res , 95 ( D13 ) : 22455 – 22462 .
- Cleveland , R B , Cleveland , W S , McRae , J E and Terpenning , I . 1990 . STL: A seasonal-trend decomposition procedure based on Loess . J Off Stat , 6 ( 1 ) : 3 – 73 .
- Cunnold , D M , Steele , L P , Fraser , P J , Simmonds , P G , Prinn , R G , Weiss , R F , Porter , L W , O'Doherty , S , Langenfelds , R L Krummel , P B . 2002 . In situ measurements of atmospheric methane at GAGE/AGAGE sites during 1985–2000 and resulting source inferences . J Geophys Res , 107 ( D14 ) doi: 10.1029/2001JD001226
- Dlugokencky , E J , Houweling , S , Bruhwiler , L , Masarie , K A , Lang , P M , Miller , J B and Tans , P P . 2003 . Atmospheric methane levels off: Temporary pause or a new steady-state? . Geophys Res Lett , 30 ( 19 ) : 1992
- Dlugokencky , E J , Bruhwiler , L , White , J WC , Emmons , L K , Novelli , P C , Montzka , S A , Masarie , K A , Lang , P M , Crotwell , A M Miller , J B . 2009 . Observational constraints on recent increases in the atmospheric CH4 burden . Geophys Res Lett , 36 : L18803
- Dlugokencky , E J , Masarie , K A , Lang , P M , Tans , P P , Steele , L P and Nisbet , E G . 1994a . A dramatic decrease in the growth rate of atmospheric methane in the northern hemisphere during 1992 . Geophys Res Lett , 21 ( 1 ) : 45 – 48 .
- Dlugokencky , E J , Masarie , K A , Lang , P M and Tans , P P . 1998 . Continuing decline in the growth rate of the atmospheric methane burden . Nature , 393 ( 6684 ) : 447 – 450 .
- Dlugokencky , E J , Myers , R C , Lang , P M , Masarie , K A , Crotwell , A M , Thoning , K W , Hall , B D , Elkins , J W and Steele , L P . 2005 . Conversion of NOAA atmospheric dry air CH4 mole fractions to a gravimetrically prepared standard scale . J Geophys Res , 110 : D18306
- Dlugokencky , E J , Steele , L P , Lang , P M and Masarie , K A . 1994b . The growth rate and distribution of atmospheric methane . J Geophys Res , 99 ( D8 ) : 17021 – 17043 .
- Esler , M B , Griffith , D WT , Wilson , S R and Steele , L P . 2000 . Precision trace gas analysis by FT-IR spectroscopy. 1. Simultaneous analysis of CO2, CH4, N2O, and CO in air . Anal Chem , 72 ( 1 ) : 206 – 215 .
- Frankenberg , C , Warneke , T , Butz , A , Aben , I , Hase , F , Spietz , P and Brown , L R . 2008 . Pressure broadening in the 2n3 band of methane and its implication on atmospheric retrievals . Atmos Chem Phys , 8 : 5061 – 5075 .
- Griffith , D WT . 1996 . Synthetic calibration and quantitative analysis of gas-phase FT-IR spectra . Appl Spectrosc , 50 ( 1 ) : 59 – 70 .
- Lassey , K R , Etheridge , D M , Lowe , D C , Smith , A M and Ferretti , D F . 2007 . Centennial evolution of the atmospheric methane budget: what do the carbon isotopes tell us? . Atmos Chem Phys , 7 : 2119 – 2139 .
- Lowe , D C , Brenninkmeijer , C AM , Brailsford , G W , Lassey , K R , Gomez , A J and Nisbet , E G . 1994 . Concentration and 13C records of atmospheric methane in New Zealand and Antarctica: evidence for changes in methane sources . J Geophys Res , 99 ( D8 ) : 16913 – 16925 .
- Lowe , D C , Brenninkmeijer , C AM , Tyler , S C and Dlugokencky , E J . 1991 . Determination of the isotopic composition of atmospheric methane and its application in the Antarctic . J Geophys Res , 96 : 15445 – 15467 .
- Lowe , D C , Manning , M R , Brailsford , G W and Bromley , A M . 1997 . The 1991–1992 atmospheric methane anomaly: southern hemisphere 13C decrease and growth rate fluctuations . Geophys Res Lett , 24 ( 8 ) : 857 – 860 .
- Mak , J E , Manning , M R and Lowe , D C . 2000 . Aircraft observations of δ13C of atmospheric methane over the Pacific in August 1991 and 1993: evidence of an enrichment in 13CH4 in the southern hemisphere . J Geophys Res , 105 ( D1 ) : 1329 – 1335 .
- Quay , P D , Stutsman , J , Wilbur , D , Snover , A , Dlugokencky , E J and Brown , T . 1999 . The isotopic composition of atmospheric methane . Global Biogeochem Cycles , 13 ( 2 ) : 445 – 461 .
- Rigby , M , Prinn , R G , Fraser , P J , Simmonds , P G , Langenfelds , R L , Huang , J , Cunnold , D M , Steele , L P , Krummel , P B Weiss , R F . 2008 . Renewed growth of atmospheric methane . Geophys Res Lett , 35 doi: 10.1029/2008GL036037
- Saueressig , G , Bergamaschi , P , Crowley , J N , Fischer , H and Harris , G W . 1995 . Carbon kinetic isotope effect in the reaction of CH4 with Cl atoms . Geophys Res Lett , 22 ( 10 ) : 1225 – 1228 .
- Saueressig , G , Crowley , J N , Bergamaschi , P , Brühl , C , Brenninkmeijer , C AM and Fischer , H . 2001 . Carbon 13 and D kinetic isotope effects in the reaction of CH4 with O(1D) and OH: new laboratory measurements and their implications for the isotopic composition of stratospheric methane . J Geophys Res , 106 ( D19 ) : 23127 – 23138 .
- Simpson , I J , Blake , D R , Rowland , F S and Chen , T-Y. 2002 . Implications of the recent fluctuations in the growth rate of tropospheric methane . Geophys Res Lett , 29 ( 10 ) doi:10.1029/2001GL014521
- Steele , L P , Dlugokencky , E J , Lang , P M , Tans , P P , Martin , R C and Masarie , K A . 1992 . Slowing down of the global accumulation of atmospheric methane during the 1980s . Nature , 358 : 313 – 316 .
- Stevens , C M and Engelkemeir , A . 1988 . Stable carbon isotopic composition of methane from some natural and anthropogenic sources . J Geophys Res , 93 ( D1 ) : 725 – 733 .
- Tans , P P . 1997 . A note on isotope ratios and the global atmospheric methane budget . Global Biogeochem Cycles , 11 ( 1 ) : 77 – 81 .
- Tyler , S C , Ajie , H O , Rice , A L , Cicerone , R J and Tuazon , E C . 2000 . Experimentally determined kinetic isotope effects in the reaction of CH4 with Cl: implications for atmospheric CH4 . Geophys Res Lett , 27 ( 12 ) : 1715 – 1718 .
- Tyler , S C , Rice , A L and Ajie , H O . 2007 . Stable isotopes in atmospheric CH4: Implications for seasonal sources and sinks . J Geophys Res , 112 : D03303 doi: 10.1029/2006JD007231
- Washenfelder , R A , Wennberg , P O and Toon , G C . 2003 . Tropospheric methane retrieved from ground-based near-IR solar absorption spectra . Geophys Res Lett , 30 ( 23 ) : 2226