Abstract
Malachite green (MG), a member of the N-methylated triphenylmethane class of dyes, has long been used to control fungal and protozoan infections in fish. MG is easily absorbed by fish during waterborne exposure and is rapidly metabolized into leucomalachite green (LMG), which is known for its long residence time in edible fish tissue. This paper describes the development of an enzyme-linked immunosorbent assay (ELISA) for the detection and quantification of LMG in fish tissue. This development includes a simple and versatile method for the conversion of LMG to monodesmethyl-LMG, which is then conjugated to bovine serum albumin (BSA) to produce an immunogenic material. Rabbit polyclonal antibodies are generated against this immunogen, purified and used to develop a direct competitive enzyme-linked immunosorbent assay (ELISA) for the screening and quantification of LMG in fish tissue. The assay performed well, with a limit of detection (LOD) and limit of quantification (LOQ) of 0.1 and 0.3 ng g−1 of fish tissue, respectively. The average extraction efficiency from a matrix of tilapia fillets was approximately 73% and the day-to-day reproducibility for these extractions in the assay was between 5 and 10%.
Introduction
Malachite green (MG) is a triphenylmethane dye that exhibits antimicrobial and antiparasitic properties (Riet et al. 2005; Yang et al. Citation2007). The use of MG has received considerable attention for its illicit use as one of the most efficacious antifungal agents in the aquaculture industry (Cha et al. Citation2001). MG is a suspected mutagen and teratogen that is easily absorbed by fish during waterborne exposure and is rapidly metabolized into leucomalachite green (LMG) in fish tissue and in water (Mitrowska et al. Citation2008). Due to the lipophilic nature of LMG, it has a long residence time in fatty tissue (t 1/2 ∼ 40 days) and has been reported to be the dominant form between these two persistent residues in fish tissue (Plakas et al. Citation1999; Andersen et al. Citation2006). This is troublesome because recent animal studies suggest that LMG has greater mutagenicity and tumorigenicity than MG (Culp et al. Citation1999; Mittelstaedt et al. Citation2004). For these reasons, MG is not approved for use as an aquaculture veterinary drug in Canada, the United States or Europe, but the worldwide misuse in aquaculture will probably continue due to its relatively low cost and availability (VRC UK Citation2003). In addition to its use as a veterinary drug, MG has been used extensively as a dye in the textile industry, as a food additive and a colouring agent (Culp et al. Citation1996). It is estimated that 20% of the dyestuff produced in the world is discharged into streams without any pre-treatment (Pourreza and Elhami Citation2007) and these sources of MG could produce potential sources for accumulation in fish tissue.
Taking into consideration the potential for intentional misuse as well as the probability of environmental contamination to waterways, surveillance of MG and LMG in aquaculture products is necessary to protect human health. Health Canada has determined that, on an interim basis, fish products containing MG or LMG residues above 1 ng g−1 will not be allowed for sale in Canada, which is more conservative than the minimum performance limit of 2 ng g−1 set by the European Union (Commission Decision Citation2004); fish with detectable MG/LMG residues below 1 ng g−1 may be allowed for sale in Canada provided that an investigation rules out intentional use of malachite green in the fish production lifecycle. Liquid chromatography (LC) and liquid chromatography–mass spectrometry (LC–MS) are two general techniques that have been used for the quantitative analysis of MG and LMG in fish tissue. These two methods have been able to detect MG and LMG with a LOD of 1.0 and 0.1 ng g−1 fish tissue, respectively (Van de Riet et al. 2005; Andersen et al. 2006). Chromatography coupled with mass spectrometry is a good technique for the analysis of toxins, but it does have some limitations. It relies on relatively expensive instruments, which need to be operated by well-trained individuals. In addition, the sample preparation that is needed to obtain such low LOD values can be very time-consuming. These aspects can sometimes be a hindrance if more rapid analysis is required to assess a shipment of product or survey a large quantity of samples, for example. Alternatively, ELISA is a rapid, specific and sensitive method that has been used for surveillance purposes to enable high throughput sample screening. There are two recent examples in the literature for the development of antibodies and immunoassays for the detection of MG and LMG (Yang et al. Citation2007; Xing et al. Citation2009). The immunogenic species developed in these two studies were prepared using a hapten synthesized from smaller aromatic building blocks, rather than LMG, resulting in the addition of a functional group directly to the unsubstituted aromatic ring of LMG. This additional functional group could potentially reduce the sensitivity of the assay because the antibodies will be derived from a hapten similar in structure to LMG rather than derived from LMG. Because LMG is recognized as the major metabolite of MG stored in fish tissue (Stammati et al. Citation2005), the present study describes the development of a hapten directly from LMG and the generation of polyclonal antibodies against this hapten to produce an ELISA to detect and quantify residues of LMG in fish.
Materials and methods
Chemicals and supplies
LMG, MG oxalate certified, leucocrystal violet (LV), crystal violet (CV), paraosaniline base (PS), ethyl chloroformate, triethylamine, iron (II) sulphate heptahydrate, m-chloroperoxybenzoic acid (m-CPBA), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDAC), N-hydroxysuccinimide (NHS), bovine serum albumin (BSA), chicken egg ovalbumin (OVA), horseradish peroxidase (HRP), human serum albumin (HSA), polyoxyethylene sorbitan monolaurate (Tween-20), hydroxylamine hydrochloride, N,N-dimetylformamide (DMF), dimethyl sulfoxide (DMSO), ethylene diamine tetraacetic acid (EDTA), glutaric anhydride, 1,4-dioxane, pyridine, chloroform, sodium tetraborate, succinic anhydride, magnesium sulphate, 2,4,6-trinitro-benzenesulfonic acid (TNBS) and Sephadex® G-100 were all purchased from Sigma-Aldrich Canada Ltd. (Oakville, Ontario, Canada). Sodium chloride, hexane, ethyl acetate, acetonitrile (ACN), and dichloromethane (DCM) were purchased from EMD Chemicals Inc (Darmstadt, Germany). Ammonium hydroxide (28–30%), p-toluene sulphonic acid (p-TSA) and anhydrous sodium acetate were purchased from BDH (Toronto, Ontario, Canada). 3,3′,5,5′-tetramethyl benzidine (TMB) substrate solutions A and B were purchased from KPL (Gaitherburg, MD, USA). Skim milk powder was purchased from Nestlé Canada Inc. and tilapia frozen skinless boneless fish fillets were purchased from a local retailer (Ottawa, Canada). The 12–14-kDa dialysis tubing was purchased from VWR Scientific (Mississauga, Ontario). Strata SCX (55 µm, 70 Å), 500 mg per 3 ml, was from Phenomenex Inc. (Torrance, CA, USA). Costar® 2592 microplates, EIA/RIA, flat bottom, certified high binding were purchased from Corning (New York, NY, USA).
Apparatus
Microplate reader: Multiskan Ascent® Thermo Labsystems (Helsinki, Finland); tissue homogenizer: Polytron® Brinkmann homogenizer, Model PT 10/35, Brinkmann Instruments Co. by Kinematika AG (Switzerland); plate shaker: Barnstead Lab-line (Melrose Park, IL, USA).
Buffers and solutions
(1) Phosphate-buffered saline (PBS): 20 mmol l−1 NaH2PO4,140 mM NaCl, pH 7.4; (2) coating buffer: 13 mmol l−1 Na2CO3 and 35 mmol l−1 NaHCO3, pH 9.6; (3) antibody dilution buffer (HSA+): 0.1% (w/v) HSA and 0.0125% (v/v) Tween-20 in PBS; (4) acetate buffer: 50 mmol l−1 CH3COONa, pH 4.5; (5) washing buffer: 0.9% NaCl (w/v), 0.0125% Tween-20 (v/v); (6) blocking buffer: 1.0% skim milk powder (w/v) in coating buffer; (7) extraction buffer: hydroxylamine solution (25% aqueous), p-toluenesulfonic acid solution (1 M), acetate buffer (8:8:84, v/v); (8) substrate solution (TMB + H2O2):(50:50, v/v); (9) assay buffer: same as antibody dilution buffer.
Synthesis of monodesmethyl-leucomalachite green (M-LMG)
LMG (0.5 g, 1.5 mmol) was dissolved in DCM (15 ml), cooled to 0°C, treated with m-CPBA (77%, 0.5 g, 2.3 mmol) in several portions and stirred for 1 h at 0°C. The reaction mixture was allowed to warm to room temperature and stirred for an additional hour. A solution of FeSO4 · 7H2O (0.840 g dissolved in 5 ml MeOH) was added and the reaction mixture was stirred at room temperature for 3 h. After this time, the solvent was evaporated under a stream of nitrogen and 45 ml of an EDTA solution (0.1 M, pH 10) was added. The product was extracted with CHCl3 (2 × 50 ml), the organic layers were dried over MgSO4, filtered and evaporated to yield a mixture of M-LMG and LMG. The mixture was purified by preparative thin layer chromatography with hexane and ethyl acetate (4:1, v/v). 1H NMR (400 MHz, CDCl3): δ 2.84 (s, 3H), δ 2.93 (s, 6H), δ 3.64 (bs, 1H), δ 5.39 (s, 1H), δ 6.56 (d, 2H, J = 8.7 Hz), δ 6.69 (d, 2H, J = 8.7 Hz), δ 6.96 (d, 2H, J = 8.7 Hz), δ 6.99 (d, 2H, J = 8.7), δ 7.16 (m, 3H), δ 7.28 (m, 2H). ESI-MS [M + H]+ = 317.
Synthesis of monodesmethyl-leucomalachite-hemiglutrate (M-LMG-HG)
M-LMG (32 mg; 0.1 mmol), was reacted with 15 mg (0.13 mmol) of glutaric anhydride in the presence pyridine (2 ml). The reaction was stirred at room temperature and the extent of the reaction was monitored using silica gel thin layer chromatography (hexane/ethyl acetate, 4:1, v/v). After the reaction was complete, the pyridine was evaporated under a stream of nitrogen and the M-LMG-HG was used without purification in the subsequent conjugation to the carrier protein. 1H NMR (400 MHz, CDCl3): δ 1.90 (m, 2H), δ 2.19 (t, 2H, J = 7.2 Hz), δ 2.37 (t, 2H, J = 7.2 Hz), δ 2.96 (s, 6H), δ 3.26 (s, 3H), δ 5.48 (s, 1H), δ 6.71 (d, 2H, J = 8.7 Hz), δ 6.99 (d, 2H, J = 8.7 Hz), δ 7.09–7.37 (m, 9H), δ 8.65 (s, 1H). ESI-MS [M + H]+ = 431.
Synthesis of monodesmethyl-leucomalachite-hemisuccinate (M-LMG-HS)
Monodes-LMG-HS was prepared similarly pathway as above using succinic anhydride.
Conjugates of monodesmethyl-leucomalachite-hemiglutrate with BSA (M-LMG-HG-BSA)
M-LMG-HG (28 mg) was dissolved in 2 ml of DMF and 1,4-dioxane (1:1, v/v) and triethylamine (10 µl) was added to the reaction mixture and stirred on ice for 10 min. Ethyl chloroformate (10 µl) was added and the reaction mixture was allowed to warm to room temperature and stirred for 1 h. This mixture was added to an ice-cold BSA solution (50 mg BSA, dissolved in 3 ml of 0.1 M sodium borate) and stirred overnight at room temperature. The final solution was dialyzed using 12–14 kDa tubing against several changes of buffer (PBS) at 4°C for 3 days, then lyophilized.
Conjugation of monodesmethyl-leucomalachite-hemisuccinate with HRP (M-LMG-HS-HRP)
M-LMG-HS (10 mg) was dissolved in DMSO (1 ml) followed by the addition of 2–3 drops of sodium tetraborate (0.1 M), EDAC (14 mg) and NHS (9 mg). The reaction mixture was incubated overnight at 4°C and then added drop wise to a solution of HRP (10 mg in 1 ml of PBS) and allowed to react at 4°C for an additional 24 h. Excess reagents were removed by dialysis against PBS for 2 days and the final HRP conjugate was concentrated using Centricon YM 30-kDa centrifugal filters (Millipore Corporation, Bedford, MA, USA) at 3500 rpm and 4°C. The concentrated conjugate was further purified by gel chromatography using Sephadex® G-100 (Sigma-Aldrich) and PBS as the mobile phase.
Antibody production
The immunization protocol and anti-serum collection were similar to those procedures described previously and are only briefly described here (Ben Rejeb et al. Citation1998). Five New Zealand white rabbits were subcutaneously inoculated with the LMG-HG-BSA conjugate in complete Freund's adjuvant. A first boost was given at day 14 after the initial inoculation and subsequent boosts were given at day 28 and continued at intervals of 28 days using incomplete Freund's adjuvant. The first anti-serum samples were taken 10 days after the third boost from the marginal vein of the ear and additional anti-serum samples were taken 10 days after subsequent boosts. In total, anti-serum was harvested 19 times per rabbit and these anti-serum samples were stored at −20°C. Trial bleeds were monitored by ELISA for their titer and the inhibition concentration corresponding to 50% response (IC50). The ex-sanguination bleeds of rabbit M484 gave the best results and the crude sera were purified using the ImmuniPure® A Plus IgG Purification Kits (Pierce Chemical Co. Rockford, IL, USA).
Assay development
Preparation of standards
LMG calibration standards ranging 0.2–50 ng ml−1 were freshly prepared in glass tubes from a master stock solution of LMG (100 µg ml−1 in methanol). A working stock solution was prepared by a 100× dilution of the master stock solution (1 µg ml−1 in 1% methanol). The final calibration standards were prepared by dilution of the working stock solution in either coating buffer or tilapia extract to give 50, 20, 8, 3.2, 1.3, 0.5 and 0.2 ng ml −1. A similar procedure was used to create a dilution series to determine the cross reactivity of MG, LV, CV and PB towards the anti-LMG antibodies.
Plate preparation for direct competitive ELISA
Microplate wells were coated with a solution of H-LMG-HG-BSA antibodies (200 µl; 1 µg ml−1 protein content in coating buffer) and allowed to incubate at 37°C for 3 h. The plate was washed five times with washing buffer and then incubated for an additional hour at 37°C in the presence of blocking buffer (200 µl) to block any additional binding sites. It should be noted that the top row of the plate was coated with only blocking buffer to determine non-specific binding. The coated plates were then washed five times with washing buffer followed by deionised water. Each wash cycle consisted of filling the wells with washing buffer, agitating the plate for a few seconds, pouring out the solution and then gently tapping the inverted microplate on absorbent tissue paper. These plates were then allowed to dry in an incubator for 15–20 min at 37°C and then stored at −20°C before use.
Immunoassay procedure
A competitive direct assay format was used for the LMG–ELISA. This procedure involves a pre-incubation step, a competition step and finally a step to develop the assay color. In a typical assay, aliquots (100 µl) of either dilution buffer, blank tilapia extract, LMG calibration standards (prepared in blank tilapia extract), or sample extracts were added in triplicate to a coated plate and pre-incubated for 1 h at 37°C with constant agitation on a plate shaker. This was followed by the addition of 100 µl of diluted HRP conjugate (1/4000) and the plate was incubated at room temperature for 30 min. After the incubation step, the wells were emptied and five wash cycles were applied. After washing, the plate was developed by the addition of 200 µl of substrate solution (TMB/H2O2; 1:1 v/v) followed by 30 min of incubation in the dark. The enzymatic colour development was stopped after 30 min by the addition of 50 µl of 6 M H2SO4 and the optical densities of each well were read 2 min later at 450 nm using a microplate reader.
Sample preparation
Spiking and extraction procedure
To determine the variability and extraction efficiency of the assay a series of spiking (fortification) studies were performed using a blank fish matrix. Tilapia samples (2.5 g) were placed into polypropylene centrifuge tubes and the appropriate volume of a working LMG solution (1 µg ml−1) was added to produce concentrations at 1.0, 12.0 and 25.0 ng g−1 of tissue. These samples were left to stand for at least 5 min at room temperature prior to extraction. The extraction of fortified tilapia samples consisted of the addition of extraction buffer (3.0 ml) and subsequent 60-s homogenization at room temperature using the tissue homogenizer. After the initial homogenization step, 10 ml of ACN was added and the sample was homogenized for an additional 60 s at room temperature. The sample was then centrifuged at 4000 rpm for 5 min and the supernatant was transferred to a clean tube. The pellet was re-extracted with 10 ml of ACN, centrifuged under the same conditions and the supernatant removed and combined with the previous supernatant fraction. DCM (5 ml) was added these combined fractions (20 ml) and shaken vigorously for 15 s followed by centrifugation at 400 rpm for 5 min. The lower organic layer was transferred into a clean borosilicate glass tube and the supernatant was extracted with an additional 5 ml of DCM. The organic layers were combined and passed through the solid phase column.
Solid phase extraction (SPE)
The Strata SCX solid phase extraction columns were first conditioned with 3 ml of conditioning solution (ACN/DCM, 80:20, v/v), then the organic layer was loaded onto the column followed by a 2-ml ACN washing, all eluted to waste. Five (5) ml of sample eluent solution (ACN/AH, 90:10, v/v) was added to the column and collected into a borosilicate glass tube. The extract was evaporated to dryness under a stream of nitrogen at 50°C, reconstituted in 1.5 ml of diluent buffer and sonicated for 10 min.
Data analysis
The blank response was calculated as the average absorbance from triplicate wells containing LMG free tilapia extracts and the non-specific binding was determined from the average absorbance of triplicate wells (coated with only blocking buffer) containing dilution buffer. A linear calibration curve was obtained from a logit (%B/B 0) versus the natural logarithm transformation of standard LMG calibration solutions,
The limit of detection (LOD) and the limit of quantification (LOQ) for this assay were determined from the average response from triplicate calibration curves. The calibration curves were produced as discussed above by spiking LMG controls into a volume of fish extract. The IC90 and the IC80 values generated from the curves were used to determine the LOD and LOQ, respectively. These values represent the absorbance at 90 and 80% of the response for the blank sample in a competitive assay, respectively.
Results and discussion
Immunogen and enzyme conjugate synthesis
The development of an immunogen containing the hapten LMG was not straightforward because the molecular structure () does not contain an active functional group, such as a hydroxyl, carboxyl or amino group to facilitate conjugation to a carrier protein. To facilitate this conjugation an alteration of the molecular structure of the hapten was necessary to provide the appropriate functionality for subsequent additions. This functionality was provided via a demethylation to convert one of the tertiary amino groups of LMG to a secondary amino group. This conversion has proven to be a convenient method of inserting functionality into a hapten that contains a tertiary amino group and has been used successfully in our laboratory for other chemicals/contaminants (unpublished results).
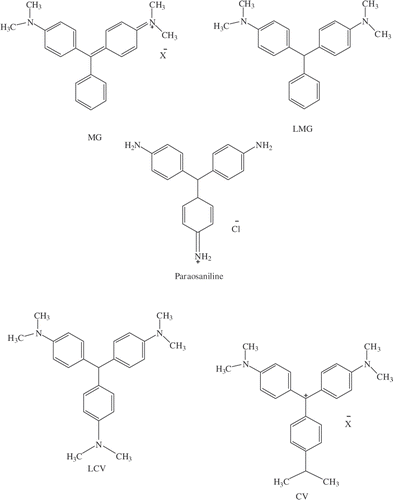
Figure 1. Structure of leucomalachite green (LMG), malachite green (MG) and structurally similar compounds, leucocrystal violet (LCV), crystal violet (CV) and paraosaniline, used to determine cross-reactivity.
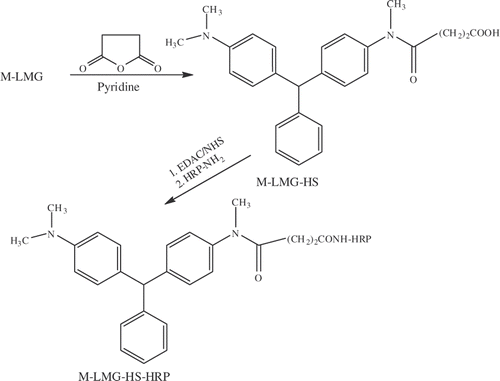
The general route to the immunogen of LMG is depicted in with the first step being the oxidative conversion of LMG into the N-oxide with m-chloroperbenzoic acid. In the next step, the N-oxide of LMG is treated with hydrated ferrous sulphate to produce M-LMG. The structure of M-LMG was confirmed by the appearance of a singlet peak in the 1H NMR (δ = 2.83) representing three protons of the N-methyl secondary amino group and a broad peak at δ = 3.24 representing the ionisable proton on this nitrogen. This is accompanied by a reduction of the integration for the singlet peak at δ = 2.93 from 12 hydrogen atoms to six. In addition, the positive ion mode electrospray ionization (ESI) mass spectrum of the product showed an intense signal at [M + H]+ = 317. M-LMG was then treated with glutaric anhydride to add a carboxylic acid group to the hapten, which could then be coupled to the carrier protein using standard techniques. The M-LMG hemiglutarate (M-LMG-HG) was confirmed by 1H NMR and the appearance of two triplets, one at δ = 2.19 and the other at δ = 2.37, and a multiplet at δ = 1.90 representing the three new CH2 groups in the molecule. The NMR assignment was confirmed by an intense signal at [M + H]+ = 431 using electrospray ionization mass spectrometry. Previous examples for the production of antibodies for LMG have relied on synthesizing a hapten from smaller building blocks, which has resulted in the addition of an extra functional group to the unsubstituted aromatic ring of LMG (Yang et al. Citation2007; Xing et al Citation2009). It appears this method has been able to produce antibodies with similar sensitivity to the ones developed in this study, but the method to develop the hapten and enzyme conjugates for this study would be less time consuming. In addition, this method to convert a tertiary amino group to a secondary amino group will be a universal process that will not rely on the reactivity of smaller building blocks and multiple synthetic processes to develop a hapten. The M-LMG-HG was conjugated to BSA via mixed anhydride method and the molar ratio of hapten per carrier protein was estimated by measuring the free amino group of the lysine side chains after reaction with TNBS. (Snyder et al. Citation1975) The differential absorption of the native and the conjugated proteins at 420 nm allowed the determination of an average conjugation ratio. In an analogous reaction, an enzyme conjugate was prepared by coupling M-LMG-HS to HRP (), which was then used to develop a competitive ELISA for LMG.
Figure 2. Synthetic scheme for the synthesis of LMG hapten and subsequent formation of immunogen by conjugation to a carrier protein (BSA).
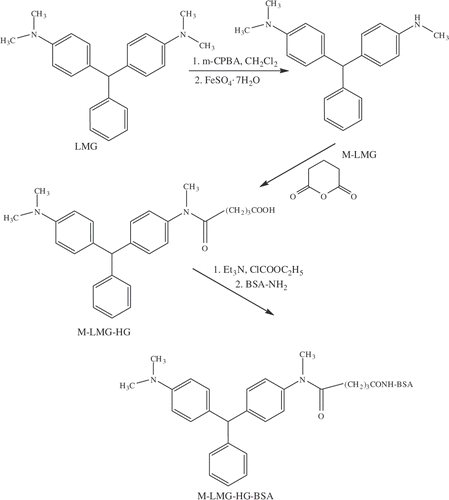
Figure 3. Synthetic scheme for the conjugation of LMG hapten to HRP.
ELISA development and optimization
In a typical development protocol, trial bleeds from rabbits immunized with M-LMG-HG-BSA were monitored for their titer using a direct ELISA format to determine the lowest IC50 value. The direct competitive ELISA format developed in this study was used for determining both the titer and the concentration of LMG in spiked samples. In this format, the M-LMG-HG-BSA antibodies are coated on the plate and then LMG and M-LMG-HS-HRP compete for available sites. The preliminary results with the optimization of all reagents in the assay, using crude sera, were promising, with IC50 values of 20 ng ml−1. Purification of the M-LMG-HG-BSA antibodies and the introduction of a pre-incubation step with LMG standard, or sample, before the introduction of the enzyme conjugate resulted in a further optimization of the IC50 value to approximately 5 ng ml−1. The sensitivity of the assay could not be optimized further using conventional ELISA techniques, but the introduction of a SPE extraction and concentration step resulted in a further decrease in the IC50 value to approximately 2.0 ng ml−1. It should also be noted that these results were determined from control samples that were prepared in a fish extract.
Assay performance
The sensitivity of the assay and its false positive rate depends on the ability to detect samples against a background response. This background response was determined by measuring the absorbance for six replicate blank tilapia extracts. The absorbance readings for these samples were all above the IC90 (0.11 ± 0.03 ng g−1 of fish tissue) value obtained from the calibration curves, which typically represents the LOD of the assay. The calibration curve consists of a zero and seven LMG controls ranging 0.1–25 ng g−1 of fish tissue (). These controls were prepared in tilapia extract to avoid any matrix effects that could complicate the interpretation of the assay. Given in are the results for the IC50, IC80 an IC90, which were produced from calibration curves generated on three different days. The results were consistent over the three days and calibration curves showed good linearity (data no shown) resulting in LOD (IC90) and LOQ (IC80) values for the assay to be 0.1 and 0.3 ng g−1 of fish tissue, respectively. To determine the extraction efficiency, repeatability and the stability of these extracts over time, fortified samples were prepared and tested over a 3-day period. Samples of tilapia (2.5 g) were fortified with the appropriate volume of working stock solution to produce concentrations of 1.0, 12.0 and 25.0 ng of LMG per gram of fish tissue. The samples were extracted using the protocol described in the experimental section and the extracts were analyzed over a 3-day period. The average results over this period for the 1.0, 12.0 and 25.0 ng g−1 fortification levels were 0.76 ± 0.08, 8.7 ± 0.8 and 17.4 ± 0.9 ng g−1, respectively. The results of this study show good recoveries and range from 70 to 76% for the extraction of LMG in fish tissue (tilapia). The results also showed that the repeatability for this ELISA is very good and ranged from 5 to 11%. The multiple day study also showed that some of the important assay components, like the antibodies, enzyme conjugate stock solutions and the extracted samples, are stable over this time frame.
Figure 4. Representative calibration curve for LMG prepared in blank tilapia extract.
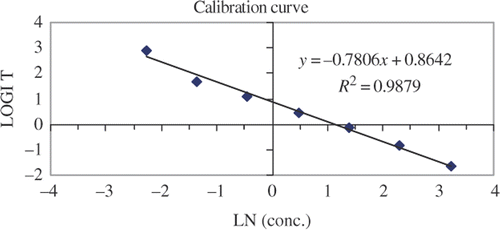
Table 1. Calibration curve results over a 3-day period with tilapia extract IC50, IC80 and IC90 in ng (LMG) g−1 tissue.
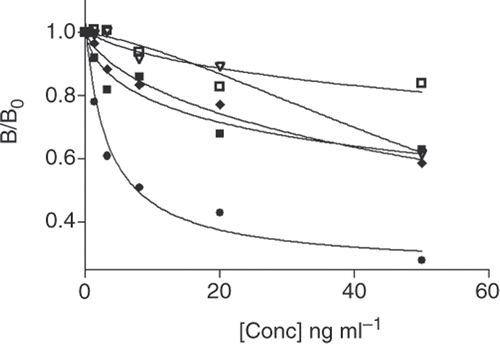
The ability of the ELISA to distinguish between LMG and compounds that are similar in structure is an important aspect that needs to be determined and can be assessed by the cross reactivity of these compounds in the assay. We tested four compounds that are similar in structure to LMG and these are shown in . To determine the full extent of the cross reactivity, the results were obtained prior to the introduction of the SPE step to remove any discrepancies that may arise due to the difference in hydrophobicity between these compounds and LMG. In a typical experiment, controls were prepared and standard curves were generated for these compounds to determine the IC50 values for each. These IC50 values were then extrapolated from the data and used to determine the % CR using the equation described in the experimental section. Malachite Green showed the highest cross-reactivity of 12%, while the other molecules investigated showed lower cross-reactivities: leucocrystal violet (2.4%), crystal violet (0.8%) and paraosaniline base (3.9%) ().
Figure 5. Calibration curves used to determine the cross-reactivity for LMG (•), MG (▪), LCV (♦), CV (□) and PS (▽).
Conclusion
A cost-effective and relatively fast SPE-ELISA method was developed for the detection and quantification of low levels of LMG in fish tissue. An easy one-pot synthetic method has been used to add functionality to a hapten that had none originally. The antibodies have been used to produce a sensitive assay with an LOD and LOQ of 0.1 and 0.3 ng g−1 of fish tissue, respectively. This ELISA method has sufficient sensitivity to enforce the maximum interim level Health Canada has set for MG and LMG of 1.0 ng g−1 of fish tissue. This assay could be used to screen large quantities of fish to detect and quantify for the presence of MG and LMG. Future work is underway to optimise the assay to screen a variety of different types of fish for LMG and MG in conjunction with LC-MS/MS analysis.
Acknowledgements
The authors would like to thank Health Canada's Bureau of Chemical Safety for funding this project. In addition, the authors thank Eric Braekevelt and Adam Becalski for their helpful comments during the editing of this manuscript.
References
- Andersen , WC , Turnipseed , SB and Roybal , JE . 2006 . Quantitative and confirmatory analyses of malachite green and leucomalachite green residues in fish and shrimp . J Agric Food Chem , 54 : 4517 – 4523 .
- Ben Rejeb , S , Fischer-Durand , N , Martel , A , Le Goffic , F , Lawrence , JF , Yeung , JM and Abbott , MA . 1998 . Development and validation of an indirect enzyme immunoassay for the detection of the herbicide isoproturon in water matrices . Int J Environ Anal Chem , 69 : 13 – 30 .
- Cha , CJ , Doerge , DR and Cerniglia , CE . 2001 . Biotransformation of malachite green by the fungus Cunninghamella elegans . Appl Environ Microbiol , 67 : 4358 – 4360 .
- Commission Decision 2004/25/EC, as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin. Off J Eur Union. L6: 38–39
- Culp , SJ and Beland , FA . 1996 . Malachite green: a toxicological review . J Am Coll Toxicol , 15 : 219 – 238 .
- Culp , SJ , Blankenship , LR , Kusewitt , DF , Doerge , DR , Mulligan , LT and Beland , FA . 1999 . Toxicity and metabolism of malachite green and leucomalachite during short-term feeding to Fischer 344 rats and B6C3F1 mice . Chem-Biol Interact , 122 : 153 – 170 .
- Mitrowska , K , Posyniak , A and Zmudzki , J . 2008 . Determination of malachite green and leucomalachite green residues in water using liquid chromatography with visible and fluorescence detection and confirmation by tandem mass spectrometry . J Chromatogr A , 1207 : 94 – 100 .
- Mittelstaedt , RA , Mei , N , Webb , PJ , Shaddock , JG , Dobrovolsky , VN , McGarrity , LJ , Morris , SM , Chen , T , Beland , FA Greenlees , KJ . 2004 . Genotoxicity of malachite green and leucomalachite green in female Big Blue B6C3F1 mice . Mutat Res , 561 : 127 – 138 .
- Plakas , SM , Doerge , DR and Turnipseed , SB . 1999 . Disposition and metabolism of malachite green and other therapeutic dyes in fish. Xenobiotics in fish , 149 – 166 . New York : Kluwer Academic/Plenum Publisher; .
- Pourreza , N and Elhami , Sh . 2007 . Spectrophotometric determination of malachite green in fish farming water samples after cloud point extraction using non-ionic surfactant Triton X-100 . Anal Chim Acta , 596 : 62 – 65 .
- Snyder , SL and Sobocinski , PZ . 1975 . An improved 2,4,6-trinitro-benzenesulfonic acid method for the determination of amines . Anal Biochem , 64 : 284 – 288 .
- Stammati , A , Nebbia , C , Angelis , ID , Albo , AG , Carltti , M , Rebecchi , C , Zampaglioni , F and Dacasto , M . 2005 . Effects of malachite green (MG) and its major metabolite, leucomalachite green (LMG), in two human cell lines . Toxicol in Vitro , 19 : 853 – 858 .
- Van de Riet , JM , Murphy , CJ , Pearce , JN , Potter , RA and Burns , BG . 2005 . Determination of malachite green and leucomalachite green in a variety of aquacultured products by liquid chromatography with tandem mass spectrometry detection . J AOAC Int , 88 : 744
- VRC UK. 2003. Veterinary Residues Committee's Annual Report on surveillance for Veterinary Residues in Food in UK for 2001, 2002, and 2003. Available from: http://www.vet-residues-committee.gov.uk
- Xing , W , He , L , Yang , H , Sun , C , Li , D , Yang , X , Li , Y and Deng , A . 2009 . Development of a sensitive and group-specific polyclonal antibody-based enzyme-linked immunosorbent assay (ELISA) for detection of malachite green and leucomalachite green in water and fish . J Sci Food Agric , 89 : 2165 – 2173 .
- Yang , MC , Fang , JM , Kuo , TF , Wang , DM , Huang , YL , Liu , LY , Chen , PH and Chang , TH . 2007 . Production of antibodies for selective detection of malachite green and the related triphenylmethane dyes in fish and fishpond water . J Agric Food Chem , 55 : 8851 – 8856 .