
ABSTRACT
In 2013 the Dutch authorities issued a warning against a dietary supplement that was linked to 11 reported adverse reactions, including heart problems and in one case even a cardiac arrest. In the UK a 20-year-old woman, said to have overdosed on this supplement, died. Since according to the label the product was a herbal mixture, initial LC-MS/MS analysis focused on the detection of plant toxins. Yohimbe alkaloids, which are not allowed to be present in herbal preparations according to Dutch legislation, were found at relatively high levels (400–900 mg kg–1). However, their presence did not explain the adverse health effects reported. Based on these effects the supplement was screened for the presence of a β-agonist, using three different biosensor assays, i.e. the validated competitive radioligand β2-adrenergic receptor binding assay, a validated β-agonists ELISA and a newly developed multiplex microsphere (bead)-based β-agonist assay with imaging detection (MAGPIX®). The high responses obtained in these three biosensors suggested strongly the presence of a β-agonist. Inspection of the label indicated the presence of N-isopropyloctopamine. A pure standard of this compound was bought and shown to have a strong activity in the three biosensor assays. Analysis by LC-full-scan high-resolution MS confirmed the presence of this ‘unknown known’ β3-agonist N-isopropyloctopamine, reported to lead to heart problems at high doses. A confirmatory quantitative analysis revealed that one dose of the preparation resulted in an intake of 40–60 mg, which is within the therapeutic range of this compound. The case shows the strength of combining bioassays with chemical analytical techniques for identification of illegal pharmacologically active substances in food supplements.
Introduction
Regulatory authorities carry out monitoring and surveillance programmes in order to ensure consumer (food) safety. This monitoring heavily relies on chemical methods that detect specific target compounds based on their physical and chemical properties. However, these chemical analytical methods most often overlook new compounds and novel hazards (‘you only find what you are looking for’). Despite the rapid developments in mass spectrometric (MS) methods, like LCxLC and hybrid-MS techniques, enabling untargeted analysis at low levels, the software and know-how to search for unexpected and unknown toxicants are still missing to a large extent. This might be tackled in future by combinations of mass databases with effect or toxicological databases, but such smart databases are still under development and will in principle still only include known substances with known toxicological effects.
Effect-based assays offer the opportunity to overcome the present difficulties and shortcomings when using chemical analytical methods alone. Such assays are able to detect both known and unknown compounds with both known and, depending on the type of assay, also yet unknown activities. Such effect-based assays are thus very useful as an early-warning system for potential hazards and suited for a risk-based food-monitoring approach as requested by the European Commission in order to protect consumers (see http://www.efsa.europa.eu/en/topics/topic/meatinspection.htm). However, effect-based assays are not able to identify the responsible compounds and ideally are combined with chemical analytical methods to confirm the presence of a regulated substance or to identify an unknown compound causing the effect. Effect-based assays and chemical methods are thus complementary and the added value of the combination has often been demonstrated (Bovee et al. Citation2006; Nielen et al. Citation2003; Bovee & Pikkemaat Citation2009; Rijk et al. Citation2009; Tooriaans et al. Citation2010).
Effect-based screening methods are very useful in the area of growth promoters, where there is the incentive to develop novel compounds for the treatment of diseases, but which can also be misused to circumvent control on the illegal application of such substances (Nielen et al. Citation2003, Citation2006). Besides the nuclear receptors, the β2-adrenergic receptor is the main target and receptor type that is responsive to anabolic agents that are illegally used in sports doping and meat production. Agents like clenbuterol, mabuterol, salmeterol and ractopamine are the best-known examples (Meenagh et al. Citation2001). Boyd et al. (Citation2009) developed and validated a competitive radioligand β2-adrenergic receptor binding assay for the detection of a broad range of β-agonists in feed. The method was validated according to EC Decision 2002/657 and proved capable of detecting 250 ng clenbuterol equivalents g–1 of sample (Boyd et al. Citation2009). This is well below the quantities normally associated with β-agonist medicated feeds. Nielen et al. used this competitive β2-adrenergic receptor binding assay to follow-up a ‘false-positive’ immunoassay result and reported the finding of an unknown β2-agonist (Nielen et al. Citation2003).
Many immunoassays have been developed for the screening of β2-agonists. For example, in the on-site tube enzyme immunoassay developed by Haasnoot et al. (Citation1996), a mixture of antibodies raised against clenbuterol and salbutamol was used, resulting in a test that showed sensitivity towards a whole range of β2-agonists. In the case of β2-agonists, antibody-based tests are suited for broad screening, as β2-agonists have a similar structure, i.e. are structurally related, and will often be recognised by the same antibody. As a result antibody-based tests are nowadays still used in monitoring programmes and enforcement approaches in the control of veterinary production.
Food supplements appear to be increasingly popular, based on the interest of consumers to use natural substances to improve health and performance. However, these supplements are regularly fortified with pharmacologically active substances to meet expectations, but in some cases can also lead to unwanted effects (Tooriaans et al. Citation2010; Reeuwijk et al. Citation2013). In 2013 the Dutch authorities issued a warning against a dietary supplement that was linked to 11 reported adverse reactions, including heart problems and in one case a cardiac arrest. In the UK a 20-year-old woman, said to have overdosed on this supplement, died. In the Netherlands the supplements were being sold in web shops and the Netherlands Food and Consumer Products Safety Authority (NVWA) put a ban on the supplement, ordering the web shops to remove the products. First analysis by LC-MS/MS in our laboratory was focused on the presence of plant toxins together with some stimulants, and revealed the presence of relatively high levels of yohimbe alkaloids. Under Dutch law, yohimbe alkaloids are prohibited in herbal preparations and this violation, together with the reported adverse effects, was serious enough to remove the product from the web shops. However, the exact cause of the adverse effects was still uncertain and the NVWA promised the public that it would elicit the exact cause of the problem. The supplement was advertised for intense energy, mental focus, reduced appetite, cravings and an anabolic state to preserve muscle. Based on this and especially the severe heart problems, it was decided to screen the supplement for the presence of a β-agonist by three different kinds of biosensor assays, i.e. a validated competitive β2-adrenergic receptor binding assay, a validated β-agonist ELISAs and a newly developed multiplex microsphere (bead)-based β-agonist assay with imaging detection (MAGPIX®) format. The present paper will show that a high response was obtained in all three biosensors confirming our suspicion of the potential presence of a β-agonist. Inspection of the label indicated the presence of N-isopropyloctopamine. A pure standard of this compound was bought and shown to have a strong activity in the three biosensor assays. In parallel, analysis by LC-full-scan high-resolution MS confirmed the presence of high amounts of this ‘unknown known’ β3-agonist N-isopropyloctopamine, reported to lead to heart problems at high doses.
Materials and methods
Chemicals
3H-dihydroalprenolol (TRK649-1 mCi 37 MBq) was obtained from Amersham Pharmacia Biotech. UK Ltd (Buckinghamshire, UK). Disodium hydrogen phosphate, sodium dihydrogen phosphate, sodium hydroxide, sodium chloride, sodium acetate, acetic acid, formic acid, ammonium formate, ammonia 32%, chloroform and ortho-phosphoric acid were obtained from Merck (Darmstadt, Germany). Sodium carbonate (Na2CO3), sodium hydrogen carbonate (NaHCO3), sodium dihydrogen phosphate monohydrate (NaH2PO4.H2O), sodium phosphate (Na2HPO4), potassium phosphate (K2HPO4), Tween-20 and sodium azide (NaN3) were obtained from VWR International (Amsterdam, the Netherlands). N-ethyl-N′-(3-(dimethylamino)propyl)carbodiimide (EDC) and N-hydroxysulphosuccinimide sodium salt (NHS) were from Pierce (Rockford, IL, USA). Clenbuterol, isoxsuprine, salmeterol, ovalbumin (OVA) and bovine serum albumin (BSA) were obtained from Sigma-Aldrich (Zwijndrecht, the Netherlands). Methanol, ethyl acetate, acetonitrile and acetone were obtained from Biosolve (Dieuze, France). Deterenol HCl (or N-isopropyloctopamine) was purchased from TLC PharmaChem., Inc. (Vaughan, Canada). Ractopamine was obtained from Bosche Scientific (New Brunswick, NJ, USA) and zilpaterol was purchased from the EU Reference Laboratory Berlin (Germany). Certify BondElut SPE cartridges (300 mg, 10 ml) were purchased from Varian (Harbor City, CA, USA). Microcon centrifugal filter devices (Ultracel YM-30) were obtained from Millipore (Darmstadt, Germany). Optiphase HI Safe 3 scintillation fluid and liquid scintillation vials were obtained from Perkin Elmer (Waltham, MA, USA). Goat-anti-rabbit–R-phycoerythrin conjugate (GAR-PE) was from Molecular Probes (Leiden, the Netherlands) and rabbit-anti-sheep–R-phycoerythrin conjugate (RAS-PE) from Santa Cruz Biotech (Heidelberg, Germany). Antibodies and HRP conjugates for the ractopamine, isoxsuprine, salmeterol and zilpaterol assay were obtained from Randox (Crumlin, County Antrim, UK) and the clenbuterol antibody was produced in-house (Haasnoot et al. Citation1996). The carboxylated microspheres numbers 012, 029, 043, 055 and 073 (MagPlex® (paramagnetic) beads), in stock bead suspensions of 1.25 × 107 beads ml–1 and sheath fluid were purchased from Luminex (Austin, TX, USA).
Sample preparation for the competitive β2-adrenergic receptor binding assay and the Luminex assay for β-agonists
Tablets were ground and homogenised to powder using a mortar and portions (1 g) of ground material were weighed into 50 ml Greiner tubes. Duplicates were fortified at 500 ng clenbuterol g–1. Samples were extracted as described previously for the validated competitive radioligand β2-adrenergic receptor binding assay (Boyd et al. Citation2009). In short, 10 ml 0.2 M phosphoric acid/methanol 1/1 (v/v) were added to the samples and controls. The samples were extracted head over head for 30 min and subsequently centrifuged for 10 min at 2700g. A 2.0 ml portion of the supernatant was transferred into a 15 ml glass tube and 5.0 ml 0.25 M sodium acetate buffer pH 4.8 were added. The extract was homogenised on a vortex and the pH was checked using indicator strips, and adjusted to pH 4.8 ± 0.2 using a acetic acid or sodium hydroxide solution when necessary. Certify BondElut columns were conditioned by applying 3 ml methanol, followed by 3 ml sodium acetate buffer (0.25 M, pH 4.8). The extracts were loaded on the columns and the columns were vacuum dried for 5 min. The columns were washed with 1 ml 1.0 M acetic acid, 6 ml methanol and 2 ml of acetone/chloroform 1/1 (v/v), having 5-min drying steps in between. The β-agonists were eluted with 7.5 ml freshly prepared 32% ammonia/ethyl acetate 3/97 (v/v). Collected fractions were evaporated gently under nitrogen at 40 ± 4°C. Residues were redissolved in 50 µl acetonitrile and diluted with 450 µl PBS buffer (0.1 M).
Sample preparation for the β-agonist ELISA
Tablets were ground and homogenised to powder using a mortar and 1 g portions of grounded material were weighed in triplicate into 14 ml test tubes. One test portion was directly used, one test portion was fortified with 0.25 ng g–1 clenbuterol and one test portion was fortified with 0.25 ng g–1 salbutamol.
Samples were extracted head over head for 30 min with 10 ml 0.2 M phosphoric acid/methanol 1/1 (v/v). After centrifugation, 1.0 ml of the upper layer was transferred into a glass tube. Each sample was also diluted 10× and 100× with 0.2 M phosphoric acid/methanol 1/1 (v/v), as high concentrations of β-agonists were expected, and the pH of each sample and each diluted sample was adjusted to pH 7.0 ± 0.5 using 8 M sodium hydroxide or 8 M acetic acid. Next, 250 µl of each sample were transferred into a new tube and 1 ml PBS with 0.05% Tween-20 (PBST) was added, samples were vortexed and centrifuged, and 250 µl were transferred into the ELISA dilution well plate.
Sample preparation for LC-MS analysis
Tablets were ground and 2.5 g of the homogenised sample was weighed into a polypropylene tube. Samples were extracted head over head for 30 min with 10 ml of water/acetonitrile 20/80 (v/v) containing 0.1% of formic acid. Tubes were centrifuged at 3500 rpm for 10 min and an aliquot of the supernatant extract was diluted 1:1 with water/acetonitrile 80/20 (v/v) containing 0.1% of formic acid and filtered (0.45 µm filter), resulting in an extract containing 0.125 g sample equivalent ml–1. For screening purposes the extract was analysed as such, but also after an additional 10- and 100-fold dilution with water/acetonitrile 50/50 (v/v) containing 0.1% formic acid.
LC-MS analysis
LC-full-scan high-resolution MS (screening)
The LC-HRMS system consisted of an Accela HPLC system coupled with a HESI II electrospray source to a single-stage Orbitrap MS (Exactive, Thermo Fisher Scientific, San Jose, CA, USA). A total of 5 μl of the extract were injected into the LC-HRMS system. The tray of the autosampler vials was maintained at 10°C. For LC separation, a 100 mm × 3 mm ID, 3 µm Atlantis T3 column from Waters (Milford, MA, USA) was used. The LC mobile phases were water (A) and methanol/water 95/5 (v/v) (B) both containing 2 mM ammonium formate and 0.002% formic acid. The LC eluent gradient was as follows: 1 min isocratic at 100% A, then a linear gradient to 55% B at 3 min and a linear gradient to 100% B at 9 min. For complete elution of all matrix co-extractants from the column, the final composition was held for 11 min. In 0.5 min the initial conditions were restored and then equilibrated for 4.5 min before the next injection. The LC flow rate was 300 µl min–1. The temperature of the column oven was 35°C.
The electrospray source was operated in positive mode using the following parameters: electrospray voltage 2.8 kV; sheath gas 19 arbitrary units; and auxiliary gas 7 arbitrary units. The heater in the source was set to 300°C and the heated capillary in the mass spectrometer was operated at 360°C. Data were acquired by continuously alternating scan events: one without fragmentation (mass range m/z 55–1000) and one with ‘all-ion-fragmentation’ at 30 eV in the high-energy collision dissociation (HCD) cell (mass range m/z 55–1000). The resolving power for both scan events was 50 000. The scan time was 0.4 s for each event. This resulted in an overall scan rate of 1.2 Hz. The automatic gain control target was set to 3*106 ions. The other parameters for the mass spectrometer were automatically tuned to obtain the highest TIC signal. Before each batch of analysis the mass calibration of the mass spectrometer was checked and optimised by the Exactive Tune v. 1.1 software (Thermo Fisher Scientific) by direct infusion of the MSCAL5 mix from Supelco (Bellefonte, PA, USA). The LC and mass spectrometer were controlled by Xcalibur 2.2 (Thermo Fisher Scientific) software.
LC-MS/MS analysis (confirmation and quantification)
The LC-MS/MS system consisted of an injection and pump system from Shimadzu (DGU-20A3 degasser; SIL-20AC XR autosampler; LC-20AD XR pump and CTO-20AC column oven) (Shimadzu’s, Hertogenbosch, the Netherlands) and an AB Sciex QTRAP 5500 mass spectrometer equipped with an ESI source operated in positive mode (AB Sciex, Nieuwerkerk a/d IJssel, the Netherlands). A total of 5 μl of the extract were injected into the LC-MS/MS system. The tray of the autosampler vials was maintained at 10°C. For LC separation, a 100 mm × 3 mm ID, 3 µm Atlantis T3 column from Waters was used. The LC mobile phases were water (A) and methanol/water 95/5 (B) both containing 5 mM ammonium formate and 0.1% formic acid. The LC eluent gradient was as follows: 1 min isocratic at 90% A, then a linear gradient to 100% B at 8 min. For complete elution of all matrix co-extractants from the column, the final composition was held for 5.5 min. In 0.5 min the initial conditions were restored and then equilibrated for 6 min before the next injection. The LC flow rate was 400 µl min–1. The temperature of the column oven was 35°C. Measurements were performed in MRM mode. Electrospray ionisation conditions were set as follows: temperature 500°C, spray voltage 4.0 kV, GS1 60 arbitrary units, GS2 50 arbitrary units. Other acquisition details are provided in .
Table 1. MS/MS parameters for N-isopropyloctopamine.
Competitive radioligand β2-adrenergic receptor binding assay
Sample extracts were analysed by the validated competitive β2-adrenergic receptor binding assay as described previously (Boyd et al. Citation2009). In short, 100 µl sample extract (obtained following the procedure described above) were incubated with 75 µl solubilised receptor-suspension (corresponding to approximately 75 µg protein), 225 µl PBS buffer and 100 µl 3H-dihydroalprenolol (30 000 dpm) in Microcon® CFDs for 45 min at 30°C. The binding reaction was stopped by adding ice-cold PBS buffer and placing the CFDs on ice. The CFDs were centrifuged to dryness at 12 000g for 2 min at 4–6°C to remove unbound 3H-dihydroalprenolol. The CFDs were washed twice with 100 µl ice-cold PBS buffer and centrifuged to dryness. The filters were transferred into scintillation vials and soaked in 10 ml Optiphase scintillation fluid for 4 h. Radioactivity, disintegrations/min of the 3H-dihydroalprenolol label (dpm), was measured by liquid scintillation counting on a Tri-Carb 2300TR LC Analyser manufactured by Packard (GMI, Ramsey, MO, USA).
β-Agonist ELISAs
Standard ELISAs were performed using 96-well plates coated with goat anti-rabbit antibodies and incubated with samples, rabbit anti-clenbuterol or rabbit anti-salbutamol antibodies, and a peroxidase-labelled salbutamol conjugate. In short for ‘clenbuterol’, a 50 µl aliquot of the sample extract (obtained following the procedure described above) was transferred into a 96-well plate and 25 µl of a diluted salbutamol–HRP conjugate solution and 25 µl of a diluted clenbuterol antiserum solution were added to each well. Similar for ‘salbutamol’, 50 µl of the sample extract were transferred into a 96-well plate and 25 µl of a diluted salbutamol–HRP conjugate solution and 25 µl of a diluted salbutamol antiserum solution were added to each well. Plates were vortex mixed and incubated overnight at 4°C. After the incubation, each plate was washed three times with 300 µl PBST/anti-foam wash solution (5.4 mM Na2HPO4, 1.3 mM K2HPO4, 150 mM NaCl; 0.05% Tween-20; 0.004% antifoam A Emulsion; pH 7.4) and 100 µl of a tetramethylbenzidine/H2O2 substrate solution were added to each well. Colouring in the dark was performed at RT for 30 min and stopped by adding 100 µl 1 M phosphoric acid.
Standards and samples were measured by using a microplate reader (Bio-Rad, iMark, Bio-Rad Laboratories B.V., Veenendaal, the Netherlands), set at a wavelength of 450 nm. Standards of clenbuterol and salbutamol as well as spiked samples were included, i.e. the first for the calculation of clenbuterol-equivalents and salbutamol-equivalents and the latter as controls. The calculations were performed automatically using the Microplate Manager v5.2 software of the microplate reader.
Multiplex bead-based assay for β-agonists
Immobilisation of β-agonist–protein conjugates on the beads
HRP conjugates of several β2-agonists, i.e. salbutamol, isoxsuprine, ractopamine, zilpaterol and salmeterol, were coupled to MagPlex® paramagnetic carboxylated beads, and each to a unique bead set, according to a standard Luminex protocol for protein coupling and as described previously (Peters et al. Citation2014). In short, the concentrated bead suspension (1.25 × 107 beads ml–1) was mixed vigorously for 5 min on a vortex and then placed in an ultrasonic bath for 1 min at RT. For each bead coupling, 200 µl of the concentrated beads were transferred to an Eppendorf tube and placed in the magnetic separator DynaMag-2™ (Invitrogen Dynal, Oslo, Norway). Beads were allowed to settle for 3 min and the supernatant was aspirated. The beads were washed by adding 100 µl water, mixed for 1 min on a vortex, placed in the magnetic separator for 3 min and the supernatant was aspirated. The beads were then washed with 80 μl 0.1 M NaH2PO4 (pH 6.3) and subsequently activated for coupling of the conjugate with 100 μl water containing 500 μg EDC and 500 μg NHS. Activation was performed for 20 min at RT. After activation, the beads were captured in the magnetic separator and washed twice with 500 µl PBS. Final conjugate concentrations were adjusted to 125 μg ml–1 in PBS (conjugate solution). The activated beads were then resuspended in 0.5 ml of this conjugate solution and incubated for 2 h at RT (in the dark with gentle agitation). After the coupling, the conjugate solutions were removed and the beads were washed four times with OVA blocking buffer (PBS buffer containing 0.1% OVA, 0.02% Tween 20 and 0.02% of sodium azide). The beads were then resuspended in 500 µl blocking buffer and stored at 4°C before use.
Multiplex bead-based assay
The measurements were performed on a planar imaging detector MAGPIX® obtained from Luminex. Plates were incubated on a Dynatech microtiter vari-shaker (Alexandria, VI, USA). During the coupling procedures, the paramagnetic microspheres (beads) were captured using the magnetic separator. All centrifugal steps were carried out in an Eppendorf 5810 R centrifuge using the A-4-62 rotor (VWR International).
shows the principle and scheme of the multiplex bead-based assay for screening sample extracts on the presence of β-agonists. Bead suspensions, with concentrations of around 100 beads μl–1 per bead set, and antibody dilutions (as a mixture) were made in 0.1% BSA blocking buffer. The assay was then performed by adding 10 μl of the mix bead suspension, 100 μl of a 100× diluted sample extract in PBST, and 10 μl of the mix antibody solution to a well of a flat-bottom 96-well plate which was then incubated for 1 h on a microplate shaker in the dark. A calibration curve was made by using 100 µl of a mixture of 5 β-agonists, i.e. clenbuterol, isoxsuprine, ractopamine, zilpaterol and salmeterol (prepared in PBST buffer) representing samples containing 0, 25, 250 and 2500 µg kg–1.
Figure 1. (colour online) Principal and schematic presentation of the multiplex bead-based β-agonist assay.
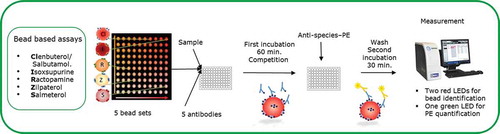
After three washing steps with PBST, 100 μl containing 600× diluted GAR-PE and 100× diluted RAS-PE was added to each well, and the plate was incubated on the microplate shaker in the dark for 30 min. After three times washing with PBST, 100 µl PBST were added to resuspend the beads. The beads were analysed with a Luminex analyser in which about 100 beads per bead set were counted. Antibody binding was quantified by the response (medium fluorescence intensity – MFI) obtained from the reporter molecule (PE) and therewith the amounts of β-agonists in the samples were quantified (taking into account that the obtained response is reciprocal to the amount of β-agonist present in the sample). The MFI is corrected for daily inter-plate fluctuations by calculating the percentage of relative binding (% B/B0), where B0 is the maximum response obtained from a sample blank.
Results and discussion
Initial targeted chemical analysis
Three batches of the supplements were obtained and initially analysed for the presence of prohibited plant toxins (pyrrolizidine alkaloids, yohimbe alkaloids, aristolochic acids) and selected stimulants (e.g. 1,3-dimethylamylamine – DMAA). As shown in , several yohimbe alkaloids were found, of which coryanthine/rauwolscine and yohimbine were at relatively high levels. Besides, as expected from the ingredient ‘Citrus Aurantium’ indicated on the label, synephrine was also found in relatively high amounts. Caffeine was present (as indicated on the labels) at high levels, but exact amounts were not quantified.
Table 2. Results of initial targeted chemical analysis by LC-MS/MS for plant toxins and selected stimulants.
The detection of yohimbe alkaloids was sufficient to withdraw the product from the market, but the determined compounds and their levels could not explain the reported adverse effects. As the supplement was advertised for intense energy, mental focus, reduced appetite, cravings and an anabolic state to preserve muscle, and because it caused severe heart problems, it was decided in addition to screen the supplement for the presence of a β-agonist.
Biosensors for screening for the presence of β2-agonists
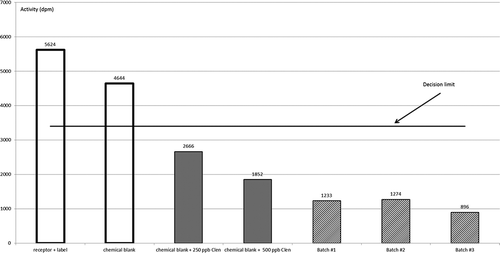
The competitive radioligand β2-adrenergic receptor binding assay was validated in our laboratory as a qualitative screening method according to the criteria in EC Decision 2002/657; it proved capable of detecting 250 ng clenbuterol equivalents g–1 of feed sample. This is well below the quantities normally associated with β-agonist-medicated feeds (Boyd et al. Citation2009). The assay is a competitive binding assay and the obtained signal is reciprocal to the amount of β-agonist present in the sample. A decision limit (CCα) of 3400 dpm was established with 20 blank feed samples. It was demonstrated that all blank samples gave a signal above the determined CCα and were thus classified as compliant or negative. All samples fortified with clenbuterol gave a signal below this CCα and were thus classified as non-compliant or suspect (Boyd et al. Citation2009). shows that two blanks, i.e. receptor plus label and a chemical blank, give signals above the established CCα and are thus classified as negative, while chemical blanks spiked with 250 or 500 ng clenbuterol per ‘g feed’ give signals below the established CCα and are thus classified as suspect. also shows that the three batches of the supplement give signals below the CCα and are thus classified as suspect. When the supplements were spiked with 500 ng clenbuterol g–1, the signals were further lowered. These results indicate that there is at least one compound or a mixture of compounds present in the supplement, and in all three batches of this supplement, that have a strong affinity for the β2-adrenoreceptor.
Figure 2. Controls and the three batches of the supplement as tested in the competitive radioligand β2-adrenergic receptor binding assay.
While this β2-adrenoreceptor competitive binding assay is used at RIKILT mainly for forensic purposes, routine monitoring and enforcement mainly relies on β-agonist ELISAs and a targeted LC-MS analysis. It was therefore decided to test the supplements also in the in-house developed and validated ‘clenbuterol’ and ‘salbutamol’ ELISAs that are frequently used to screen veterinary samples for the NVWA. These ELISAs are also competitive binding assays and the obtained signal is reciprocal to the amount of β-agonist that is present in the sample, i.e. intense colouring means no β-agonists present while light colouring indicates the presence of β-agonists. Both ELISAs are more sensitive than the β2-adrenoreceptor competitive binding assay and capable of detecting levels as low as 25 ng clenbuterol-equivalents or salbutamol-equivalents g–1. shows that the spiked blank samples lowered the mean OD value as measured at 450 nm and also shows that all supplement sample extracts and their 10- and 100-fold dilutions resulted in (near) maximal responses in both ELISAs, meaning that all supplement samples were found to be suspect for the presence of a β-agonist. The OD values of the 100-fold diluted sample extracts increased a little in the clenbuterol ELISA, while they stayed at a minimum in the salbutamol ELISA, indicating that the responsible compound(s) in the supplement has a structure more similar to that of salbutamol than clenbuterol.
Table 3. Results of controls and the three batches of the supplement as tested in the clenbuterol and salbutamol ELISAs.
Recently a multiplex screening assay for the broad detection of β-agonists was developed on a bead-based platform, similarly as developed before for coccidiostats (Bienenmann-Ploum et al. Citation2013). The microsphere-based assays are, just as the ELISAs, based on specific antibodies and also here the obtained signal is reciprocal to the amount of β-agonist present in the sample. All results are summarised in , giving the relative inhibition compared with a blank control (B/B0 = 100). The results with the standard mixture containing five different β-agonists show that an increasing concentration of the β-agonist leads to a decreasing response on the corresponding paramagnetic bead set (, samples 1–4: representing samples containing 0, 25, 250 and 2500 µg kg–1 of each of the β-agonists, i.e. clenbuterol, isoxsuprine, ractopamine, zilpaterol and salmeterol). The responses of the salbutamol/clenbuterol, isoxsuprine, ractopamine and zilpaterol beads to their corresponding β-agonists was similar, while the response by salmeterol on the salmeterol bead was less pronounced. No inhibition was observed with the chemical blank (, sample 5). The sample extracts of the three batches of the supplement resulted in clear responses on the salbutamol/clenbuterol, isoxsuprine, ractopamine and zilpaterol beads (, samples 6–8).
Figure 3. Results (% B/B0) of a β-agonist mixture (1–4), a chemical blank (5) and sample extracts of three batches of the supplement causing hart problems (6–8) tested in a newly developed multiplex bead-based β-agonist assay.
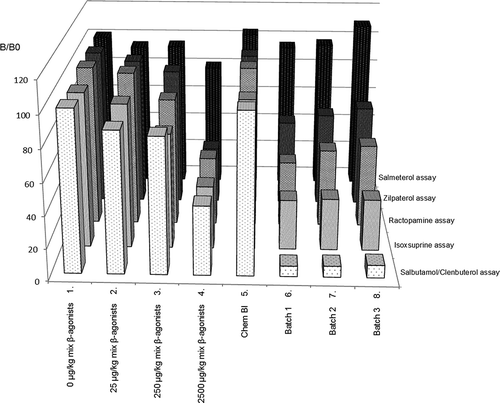
Thus, all three batches of the supplement were classified as ‘suspect’ in the three biosensor assays, i.e. the validated competitive β2-adrenergic receptor binding assay, the validated β-agonist ELISAs and the newly developed multiplex bead-based β-agonist assay. The biosensor results also indicate that the responsible β-agonist has a clear affinity for the β2-adrenergic receptor and a structure similar to that of salbutamol and clenbuterol, and especially to that of salbutamol, as the inhibition of the sample extracts was most pronounced on the salbutamol/clenbuterol bead (, samples 6–8) and in addition was more pronounced in the salbutamol ELISA than the clenbuterol ELISA ().
Chemical screening and confirmation of suspect β-agonist isopropyloctopamine
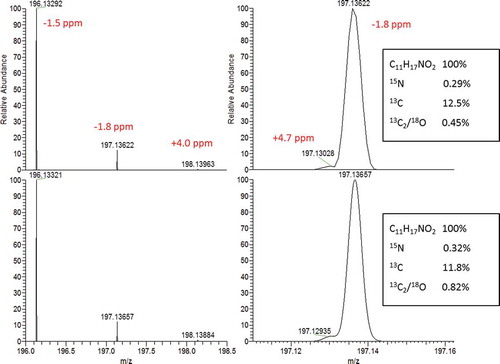
A more thorough inspection of the label on the supplement itself and on the internet then highlighted the potential presence of N-isopropyloctopamine, a known β3-agonist that can lead to heart problems at high doses. A chemical screening method based on LC with high-resolution MS was performed to investigate the presence of this compound. An intense signal for the exact mass of the protonated molecule was obtained. The accurate masses and isotope pattern matched with C11H17NO2 + H+, supporting the presence of isopropyloctopamine (). Based on this finding, an analytical standard of the pure compound, deterenol HCl which is the same as N-isopropyloctopamine, was ordered and a confirmatory quantitative analysis based on LC-MS/MS was performed. This confirmed the identity. The concentrations found in the three samples were 14, 18 and 21 mg g–1, corresponding to 39, 50 and 59 mg per daily dose (two tablets of approximately 1.4 g day–1).
Figure 4. Experimental (top) and simulated (bottom) profile mass spectrum of C11H17NO2 + H+ (resolving power 50 000 full width at half maximum). The zoomed-in detail on the right shows the 15N13C isotope cluster (15N being the shoulder on the left-hand side of the mass peak). Relative abundance of the isotopes are indicated in the boxes.
Relative ‘salbutamol activity’ in the supplements and semi-quantitative determination of the N-isopropyloctopamine amounts by the different biosensors
The pure compound N-isopropyloctopamine (IPO) was tested in the three bioassays simultaneously with the above-described chemical analytical analysis. – show the results obtained with IPO in the competitive β2-adrenergic receptor binding assay, the ‘salbutamol’ ELISA and the β-agonist Luminex method, respectively. These results show that this known β3-agonist is positively tested in these three biosensor assays. Using a semi-quantitative approach, the results as presented in indicate that IPO is about 500 times less active on the β2-adrenergic receptor compared with clenbuterol (0.2%), as the inhibiting response of a chemical blank spiked with 500 ppb clenbuterol was similar to that of a chemical blank spiked with 250 000 ppb deterenol. The results presented in also indicate that the IPO content exceeds the amount of 0.25 mg g–1, as the sample extracts resulted in higher responses than a control spiked with 0.25 mg g–1 IPO (250 000 µg kg–1).
Figure 5. Controls, IPO standards (Deterenol) and the three batches of the supplement as tested in the competitive radioligand β2-adrenergic receptor binding assay.
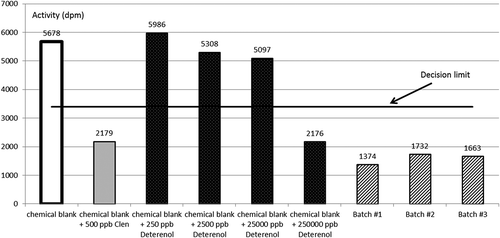
Figure 6. Response of standard series of clenbuterol (clen) in the ‘clenbuterol’ ELISA (left y-axis) and of salbutamol (salb) and IPO in the ‘salbutamol’ ELISA (right y-axis).
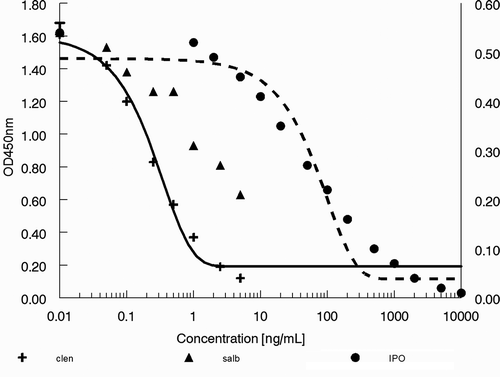
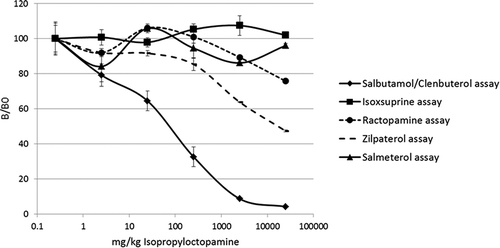
Figure 7. Results (% B/B0) of N-isopropyloctopamine (IPO) tested in a newly developed multiplex bead-based β-agonist assay for screening feed and food samples.
shows the response of the standard series of clenbuterol in the ‘clenbuterol’ ELISA and of salbutamol and IPO as determined in the ‘salbutamol’ ELISA. The 50 ng IPO ml–1 standard was calculated to contain 2.028 ng salbutamol-equivalents ml–1 and thus the cross-reactivity of IPO in this ELISA is 4%. Lower responses for IPO were obtained in the ‘clenbuterol’ ELISA (data not shown). The data in show that the 100-fold diluted sample extracts still caused a near maximal response, i.e. OD values below 0.05. These 100-fold diluted sample extracts correspond in the ELISA to 0.1 mg of the tablet per ml. shows that a signal below an OD value of 0.05 corresponds to approximately 2000 ng IPO ml–1. Based on these ELISA data, the tablets are calculated to contain about 20 mg IPO g–1, which is similar to the LC-MS/MS determined contents of 14, 18 and 21 mg g–1.
The results obtained the bead-based assays indicated that IPO shows a cross-reactivity on the salbutamol/clenbuterol bead of about 1%, as a chemical blank spiked with 250 µg kg–1 clenbuterol () resulted in a similar response as a chemical blank spiked with 25 000 µg kg–1 IPO (), while no clear responses were observed on the isoxsuprine, ractopamine, zilpaterol and salmeterol beads (). This is in agreement with the cross-reactivity as estimated in the ELISAs (4% in the salbutamol ELISA). The results presented in and also indicate that the IPO content clearly exceeds the amount of 0.25 mg g–1, as the sample extracts () resulted in higher responses than a control spiked with 0.25 mg g–1 IPO (250 000 µg kg–1) ().
Conclusions
The development of a cell-based specific reporter gene assay for β-agonists is relatively very difficult, as the β2-adrenergic receptor is located in the outer cell membrane and has no direct interaction with the DNA. Upon binding of an agonist, these seven transmembrane receptors activate an adenylate cyclase via a G-protein. The subsequent formation of the intracellular adenosine 3ʹ,5ʹ-cyclic monophosphate (cAMP) signalling molecule is not specific enough and can be influenced by many other cell processes (Bovee & Pikkemaat Citation2009). The most specific cell-based bioassay for β-agonists is the assay developed by Takeda et al. using Chinese hamster ovary (CHO) cells stably expressing E3-tagged β2-adrenergic receptors (E3-β2ARs) (Takeda et al. Citation2012). It only requires normal cell culture facilities and a fluorescence image analyser, although the latter is rather expensive.
To date, the most easy, cheap, reliably and therefore most used bio-based screening methods for β-agonists are β-adrenergic receptor binding assays and antibody-based tests. In the present study three different biosensors were used to screen three batches of a supplement that was linked to 11 reported adverse reactions in the Netherlands, including heart problems, some hospitalisations and in one case even a cardiac arrest. The outcomes confirmed and proved the suitability of these tests to detect different types of β-agonist, enabling the flagging of supplements containing unknown or undeclared ingredients with β-adrenergic activities (screened as suspect). The present study also demonstrates that combining biosensors with chemical analytical techniques results in a powerful strategy to elicit the exact cause in case of intoxications, as the biosensor outcomes direct the chemical analytical researchers to the right track, i.e. give a clear hint to what kind of compounds and masses to look for and focus on in their analysis. More generally, this case shows that natural supplements may contain plant toxins but also chemically produced drugs that present a health risk to the user, who is probably unaware of their presence. The application of effect-based assays may prevent producers to switch to (un)known substances that are often less well characterised, e.g. for their adverse effects in humans.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bienenmann-Ploum ME, Vincent U, Campbell K, Huet AC, Haasnoot W, Delahaut P, Stolker LA, Elliott CT, Nielen MW. 2013. Single-laboratory validation of a multiplex flow cytometric immunoassay for the simultaneous detection of coccidiostats in eggs and feed. Anal Bioanal Chem. 405:9571–9577.
- Bovee TFH, Bor G, Heskamp HH, Hoogenboom LAP, Nielen MWF. 2006. Validation and application of a robust yeast estrogen bioassay for the screening of estrogenic activity in animal feed. Food Addit Contam. 23:556–568.
- Bovee TFH, Pikkemaat MG. 2009. Bioactivity-based screening of antibiotics and hormones. J Chromatogr. 1216:8035–8050.
- Boyd S, Heskamp HH, Bovee TFH, Nielen MWF, Elliott CT. 2009. Development, validation and implementation of a receptor based bioassay capable of detecting a broad range of β-agonist drugs in animal feedingstuffs. Analytica Chimica Acta. 637:24–32.
- Haasnoot W, Cazemier G, Stouten P, Kemmers-Voncken A. 1996. In residue analysis in food safety: Applications of immunoassay methods. In: Beier RC, Stanker LH, editors. Immunoassays for Residue Analysis—Food Safety Washington, DC: American chemical society; p. 60–73.
- Meenagh SA, Elliot CT, Buick RK, Izeboud CA, Witkamp RF. 2001. The preparation, solubilisation and binding characteristics of a β2-adrenoceptor isolated from transfected Chinese hamster cells. Analyst. 126:491–494.
- Nielen MW, Elliott CT, Boyd SA, Courtheyn D, Essers ML, Hooijerink HH, Van Bennekom EO, Fuchs RE. 2003. Identification of an unknown beta-agonist in feed by liquid chromatography/bioassay/quadrupole time-of-flight tandem mass spectrometry with accurate mass measurement. Rapid Commun Mass Spectrom. 17:1633–1641.
- Nielen MWF, Bovee TFH, Van Engelen MC, Rutgers P, Hamers ARM, Van Rhijn JA, Hoogenboom LAP. 2006. Urine testing for designer steroids by liquid chromatography with androgen bioassay detection and electrospray quadrupole time-of-flight mass spectrometry identification. Anal Chem. 78:424–431.
- Peters J, Cardall A, Haasnoot W, Nielen MW. 2014. 6-Plex microsphere immunoassay with imaging planar array detection for mycotoxins in barley. Analyst. 139:3968–3976.
- Reeuwijk NM, Talidda A, Malisch R, Kotz A, Tritscher A, Fiedler H, Zeilmaker MJ, Kooijman M, Wienk KJH, Traag WA, Hoogenboom LAP. 2013. Dioxins (polychlorinated dibenzo-p-dioxins and polychlorinated dibenzo-furans) in traditional clay products used during pregnancy. Chemosphere. 90:1678–1685.
- Rijk JCW, Bovee TFH, Wang S, Van Poucke C, Van Peteghem C, Nielen MWF. 2009. Detection of anabolic steroids in dietary supplements: the added value of an androgen yeast bioassay in parallel with a chromatography-tandem mass spectrometry screening method. Analytica Chimica Acta. 637:305–314.
- Takeda Y, Yano Y, Matsuzaki K. 2012. Detection of oligomerization of membrane proteins using coiled-coil tag-probe labeling method and in-cell fluorescence spectroscopy. Anal Chem. 84:1754–1759.
- Tooriaans AWFT, Bovee TFH, De Rooy J, Stolker LAAM, Hoogenboom LAP. 2010. Gynaecomastia linked to the intake of an herbal supplement fortified with diethylstilbestrol. Food Addit Contam. 27:917–925.